
Introduction to Cystic Fibrosis and Airway Surface Liquid
Cystic Fibrosis (CF) is a genetic disorder that primarily affects the respiratory system, leading to chronic lung infections and progressive lung damage. The disease is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes a protein responsible for regulating the transport of chloride and bicarbonate ions across epithelial cell membranes. One of the critical consequences of CFTR dysfunction is the alteration of the airway surface liquid (ASL) pH, which plays a crucial role in maintaining the airway’s innate defence mechanisms (Figure 1).
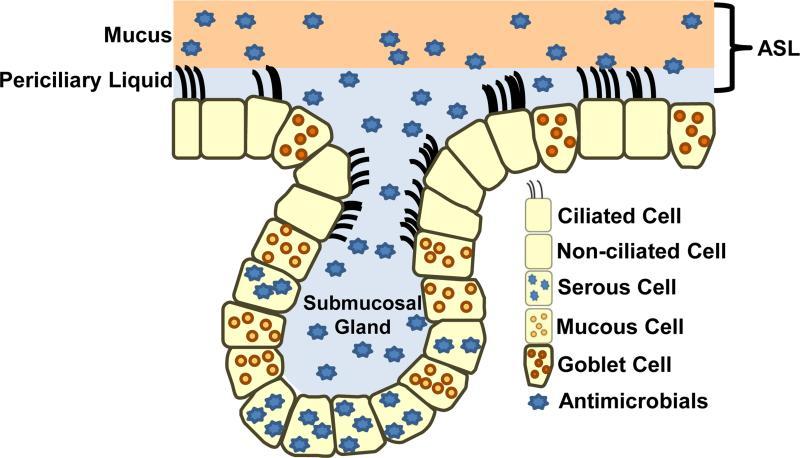
Figure 1: A simplified model of the airway epithelium. The airway epithelium has two compartments, the surface epithelium and the submucosal gland (SMG) epithelium. The airway surface epithelium includes ciliated and non-ciliated cells, goblet cells, and basal cells (not shown). SMG epithelium includes ciliated duct cells, mucus cells, and serous cells that secrete antimicrobial proteins. ASL provides a barrier between the epithelium and inspired air. ASL is composed of two layers, a mucus (gel) layer and periciliary liquid. The periciliary liquid covers the cilia, providing an environment for the beating of the cilia and ASL clearance when pathogens become trapped. Various antimicrobials are found in ASL.
The ASL is a thin layer of fluid that lines the respiratory epithelium, providing a critical interface between the airway cells and inhaled pathogens. This fluid layer consists of two components: a periciliary liquid layer, which facilitates ciliary movement and mucus clearance, and a mucus layer, which traps and removes inhaled particles and microorganisms. The pH of the ASL is tightly regulated, and even slight deviations can significantly impact the airway’s ability to defend against infections.
Role of ASL pH in Host Defense
The pH of the ASL is a key determinant of its antimicrobial properties. Under normal conditions, the ASL maintains a slightly alkaline pH, which is optimal for the activity of various antimicrobial peptides and enzymes. These molecules, such as lysozyme, lactoferrin, and defensins, play a critical role in neutralising and eliminating pathogens that enter the airways. By maintaining an alkaline pH, the ASL ensures that these antimicrobial agents function effectively, providing a robust first line of defence against respiratory infections.
In CF, the defective CFTR protein impairs the transport of bicarbonate ions, leading to a reduction in the ASL pH. This acidification of the ASL has profound effects on its antimicrobial properties. The reduced pH can inhibit the activity of antimicrobial peptides, rendering the airways more susceptible to infections. Moreover, an acidic ASL environment can promote the growth and persistence of pathogenic bacteria, such as Pseudomonas aeruginosa, which are commonly associated with chronic lung infections in CF patients.
The acidification of the ASL in CF not only compromises the airway’s innate defences but also contributes to the chronic cycle of infection and inflammation that characterises the disease. The persistent presence of pathogens in the airways leads to ongoing inflammation, further damaging the lung tissue and exacerbating the progression of CF.
Mechanisms of ASL Acidification in Cystic Fibrosis
The mechanisms underlying ASL acidification in CF are closely linked to the defective CFTR protein. The CFTR protein normally functions as a chloride and bicarbonate ion channel, playing a critical role in maintaining the ionic balance of the ASL. In CF, the loss of CFTR function disrupts this balance, leading to a decrease in bicarbonate secretion and an accumulation of protons (H+) in the ASL, resulting in a lower pH.
Several factors contribute to the acidification of the ASL in CF. Firstly, the defective CFTR protein directly impairs bicarbonate ion transport, reducing the buffering capacity of the ASL. This reduction in buffering capacity makes the ASL more prone to acidification, particularly in response to environmental challenges, such as inhaled pollutants or pathogens.
Secondly, the impaired chloride ion transport in CF leads to an overall reduction in ASL volume. The decreased ASL volume can concentrate the remaining bicarbonate ions, further contributing to acidification. Additionally, the reduced ASL volume impairs mucociliary clearance, leading to the accumulation of mucus and trapped pathogens in the airways.
Finally, the chronic inflammation associated with CF can exacerbate ASL acidification. Inflammatory cells, such as neutrophils, release acidic by-products, such as lactic acid, into the ASL. The accumulation of these acidic by-products can further lower the pH of the ASL, creating a vicious cycle of acidification, impaired host defence, and ongoing infection and inflammation.
Implications for Treatment and Therapeutic Strategies
Understanding the effects of ASL pH on host defence in CF has significant implications for the development of new therapeutic strategies. Restoring the pH balance of the ASL represents a potential therapeutic approach for enhancing the airway’s innate defences and reducing the burden of chronic lung infections in CF patients.
One promising therapeutic strategy is the use of bicarbonate therapy to directly increase the pH of the ASL. By delivering bicarbonate ions to the airways, this approach aims to counteract the acidification caused by CFTR dysfunction, restoring the optimal pH for antimicrobial activity. Preliminary studies have shown that bicarbonate therapy can improve ASL pH and enhance the activity of antimicrobial peptides, suggesting potential benefits for CF patients.
Another approach is the use of CFTR modulators, which are drugs designed to improve the function of the defective CFTR protein. CFTR modulators, such as ivacaftor and lumacaftor, have shown promise in restoring CFTR function and improving ASL pH in CF patients. By enhancing bicarbonate transport, these drugs may help restore the antimicrobial properties of the ASL and reduce the frequency and severity of lung infections.
Additionally, therapies targeting the inflammatory response in CF could also help mitigate ASL acidification. Anti-inflammatory drugs, such as corticosteroids or non-steroidal anti-inflammatory drugs (NSAIDs), may reduce the production of acidic by-products by inflammatory cells, helping to maintain a more alkaline ASL environment. Reducing inflammation may also help preserve lung function and slow the progression of CF.
Emerging research is also exploring the potential of gene therapy as a long-term solution for correcting the underlying CFTR defect. By delivering a functional CFTR gene to the airway epithelial cells, gene therapy could potentially restore normal CFTR function and prevent ASL acidification at its source. While gene therapy is still in the experimental stages, it represents a promising avenue for the future treatment of CF.
Conclusion
The pH of the airway surface liquid is a critical factor in maintaining the airway’s innate defence mechanisms. In cystic fibrosis, the defective CFTR protein leads to a reduction in ASL pH, compromising the antimicrobial properties of the ASL and increasing susceptibility to chronic lung infections. Understanding the mechanisms of ASL acidification in CF has opened new avenues for therapeutic intervention, with approaches such as bicarbonate therapy, CFTR modulators, and anti-inflammatory treatments showing promise in restoring ASL pH and enhancing host defence.
As research continues to advance, the development of therapies that target ASL pH and other aspects of CF pathophysiology holds the potential to significantly improve the quality of life for patients with cystic fibrosis. By addressing the underlying causes of ASL acidification, these therapies may help break the cycle of infection and inflammation that drives the progression of CF, offering hope for better outcomes in this challenging disease.
Journal article: Effects of airway surface liquid pH on host defense in cystic fibrosis. The International Journal of Biochemistry and Cell Biology.
Summary by Faith Oluwamakinde
References
- Saint-Criq, V., & Gray, M. A. (2017). Role of CFTR in epithelial physiology. Cellular and Molecular Life Sciences, 74(1), 93-117.
- Pezzulo, A. A., Tang, X. X., Hoegger, M. J., et al. (2012). Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature, 487(7405), 109-113.
- Zhou, Y., Song, J., & Willenbring, J. (2018). Mechanisms of Airway Surface Liquid Acidification in Cystic Fibrosis. Frontiers in Physiology, 9, 1464.
- Garvin, L. M., Chen, Y., & Gordon, W. M. (2019). The potential of bicarbonate therapy in cystic fibrosis. American Journal of Physiology-Lung Cellular and Molecular Physiology, 317(6), L899-L914.
- Mall, M. A., & Hartl, D. (2014). CFTR: cystic fibrosis and beyond. European Respiratory Journal, 44(4), 1042-1054.
- Ratjen, F., & Döring, G. (2003). Cystic fibrosis. The Lancet, 361(9358), 681-689.