





Heart failure (HF) remains a major health concern despite medical advancements. This condition often leads to repeated hospitalizations, suggesting it increases the risk of future complications and contributes to the development of multiple chronic illnesses (multimorbidity). Chronic inflammation, a hallmark of many diseases, is a common thread in multimorbidity, but how HF fuels inflammation and drives multimorbidity remains unclear.
A recent study sheds light on a surprising culprit in HF’s progression: Hematopoietic stem cells (HSCs) (Figure 1). These master cells in the bone marrow generate various blood cells, including immune cells called monocytes. Researchers explored how HF alters HSCs, their monocyte offspring, and how these changes impact the heart, muscles, and kidneys.
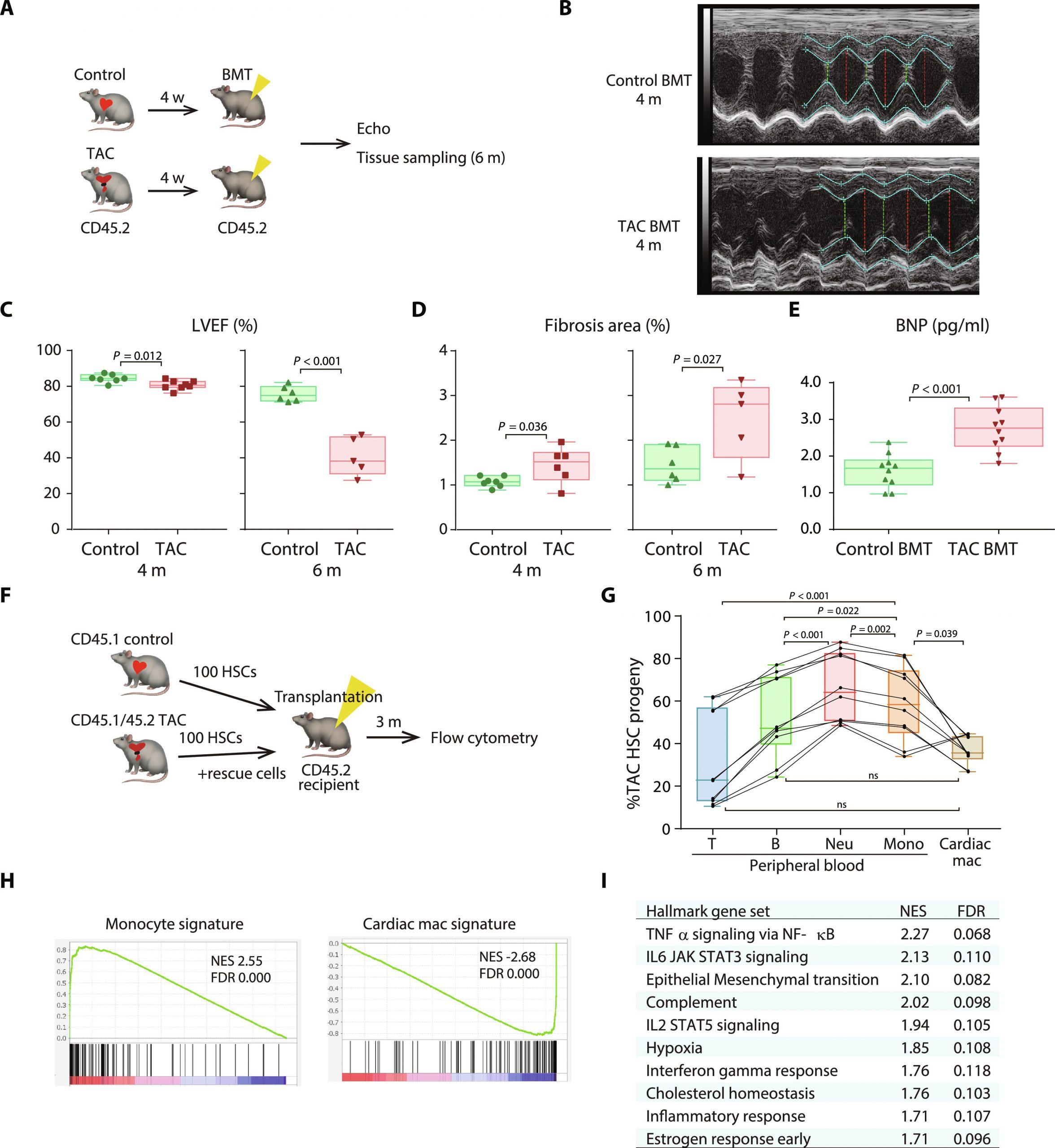
Figure 1: Transplantation of BM from TAC mice induces spontaneous cardiac dysfunction. (A) Schematic depicting the protocol for BMT from mice that have received either sham or TAC into recipient mice. (B) Representative images of M-mode echocardiography. (C) Echocardiographic assessment of left ventricular ejection fraction (LVEF) 4 or 6 months after BMT. Additional echo parameters are shown in fig. S1 (A and B). Note that, for the 4- and 6-month echo studies, two independent sets of animals were used. The results of consecutive echo studies in another set of animals are shown in fig. S1 (C and D). (D) Histological analysis of fibrotic areas identified using Picro-Sirius red staining. With the exception of two mice from the 4-month cohort that died after the echo procedure, the same mice used for the echo studies in (C) were used for the histological studies. Representative histological images are shown in fig. S1E. (E) Plasma concentrations of brain natriuretic peptide (BNP) in the recipients 6 months after BMT. (F) Schematic depicting the protocol for competitive HSC transplantation from control and 4-week-TAC mice. (G) Flow cytometric analysis of CD45+ cells in peripheral blood (left) and cardiac macrophages (right) in recipient mice 3 months after transplantation, n = 10 recipient mice. Shown are fractions of TAC HSC–derived cells among the total transplanted HSC-derived cells in each compartment. One-way repeated-measures ANOVA were conducted, followed by the post hoc Tukey-Kramer test after logit transformation. Flow cytometry gating strategy provided in fig. S2. (H) GSEA of genes differentially expressed between cardiac macrophages derived from control or TAC LSK cells 2 months after transplantation. The gene sets are composed of genes that were differentially expressed between circulating monocytes and cardiac macrophages (data file S1). Left: Gene set up-regulated in monocytes; right: up-regulated in cardiac macrophages. NES, normalized enrichment score. (I) The top 10 MSigDB hallmark gene sets significantly up-regulated in TAC-experienced BM-derived cardiac macrophages are shown. Box-and-whisker plots show the median, 5th and 95th percentiles, and the minimum and maximum values. Two-tailed unpaired Student’s t test was used in (C), (D), and (E).
The study induced HF in mice and then transplanted their bone marrow into healthy mice. These recipient mice developed impaired heart function and increased fibrosis (scarring) compared to those receiving healthy bone marrow. This suggests that HF alters HSCs, making them less effective and potentially contributing to further heart damage.
Researchers delved deeper, investigating how HF affects the development and function of monocytes derived from HSCs. They discovered that HF skews HSCs towards producing more inflammatory monocytes and fewer tissue-resident macrophages (cells crucial for tissue health). This shift towards inflammation could disrupt heart function and contribute to tissue damage.
Further analysis revealed that HF disrupts a critical signaling pathway called TGF-β, vital for HSC hibernation (a resting state). Reduced TGF-β signaling may explain the increased proliferation of HSCs observed in HF. Interestingly, treatment with TGF-β reversed this effect, suggesting its potential role in regulating HSC function and preventing HF progression.
The study highlights the bone marrow as a central player in HF’s detrimental effects. HF appears to imprint “stress memories” on HSCs, altering their function and promoting inflammation. These altered HSCs contribute not only to further heart damage but also potentially increase the risk of damage to other organs like muscles and kidneys, ultimately leading to multimorbidity.
This research opens doors for new therapeutic approaches targeting HSCs and TGF-β signaling. By mitigating the negative impact of HF on stem cells, researchers hope to disrupt the cycle of inflammation and improve outcomes for patients with this debilitating condition.
Journal article: Nakayama., Y., et al., 2024. Heart failure promotes multimorbidity through innate immune memory. Science Immunology
Summary by Stefan Botha



