





Antimicrobial peptides (AMPs) are small, naturally occurring molecules that form a crucial part of the innate immune system. They are widely distributed across different tissues and serve as the body’s first line of defence against a broad spectrum of pathogens, including bacteria, fungi, and viruses. In the respiratory tract, AMPs play a particularly important role in maintaining the integrity of the airway mucosa and preventing infections. This function is of significant interest in the context of cystic fibrosis (CF), a genetic disorder characterised by chronic respiratory infections and inflammation due to defective mucociliary clearance. This article will explore the expression and roles of antimicrobial peptides in the airway mucosa, with a focus on their potential implications in cystic fibrosis. (Figure 1)
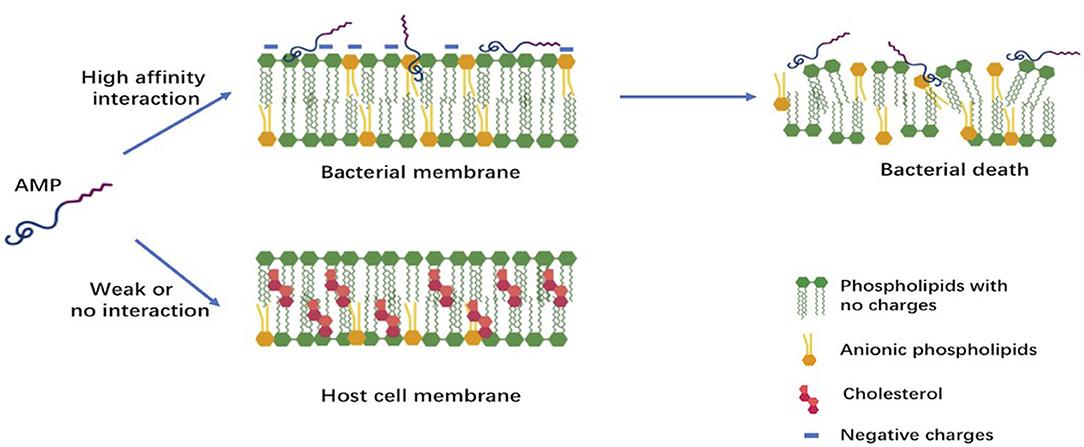
Figure 1. Early interactions of cationic antimicrobial peptides with bacterial or host cell membrane. The anionic molecules in the membranes of Gram-negative and Gram-positive bacteria attract cationic AMPs via electrostatic and hydrophobic interactions. In contrast to bacteria, the cytoplasmic membrane of host cells with a neutral net charge connects with cationic AMPs via hydrophobic interactions, which are relatively weak.
Antimicrobial Peptides in the Airway Mucosa
The airway mucosa is constantly exposed to various pathogens and environmental pollutants, making an effective defence system essential for maintaining respiratory health. AMPs are a critical component of this defence system. They are produced by epithelial cells, neutrophils, and other immune cells in the respiratory tract and act by disrupting the cell membranes of pathogens, leading to their destruction. In addition to their direct antimicrobial activities, AMPs also modulate the immune response, promoting the recruitment and activation of immune cells, and enhancing the clearance of pathogens.
Among the most studied AMPs in the airway mucosa are defensins and cathelicidins. Defensins, including α-defensins and β-defensins, are small cationic peptides that exhibit broad-spectrum antimicrobial activity. They are expressed constitutively in the airway epithelial cells and can be upregulated in response to infection or inflammation. β-defensins are induced by pro-inflammatory cytokines and are involved in recruiting immune cells to sites of infection. Cathelicidins, especially LL-37, are another class of AMPs that are expressed in the airway mucosa and are known for their potent antimicrobial and immunomodulatory activities.
Role of AMPs in Cystic Fibrosis
Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, leading to defective chloride ion transport and thick, sticky mucus in the airways. This abnormal mucus impairs mucociliary clearance, creating an environment conducive to chronic bacterial infections, particularly with pathogens like Pseudomonas aeruginosa and Staphylococcus aureus. Chronic infections, in turn, lead to persistent inflammation, which further damages the airway epithelium and exacerbates disease progression.
The role of AMPs in CF has been the subject of extensive research, as these peptides are critical in maintaining mucosal defence in the respiratory tract. However, in CF patients, the expression and function of AMPs are often compromised. Studies have shown that the ionic imbalance in CF airways, particularly the reduced levels of chloride and bicarbonate ions, can impair the antimicrobial activity of peptides like defensins. This is because the activity of many AMPs is dependent on ionic conditions, and the altered ion composition in CF airways reduces their ability to kill pathogens effectively.
Moreover, the thick mucus in CF patients can act as a physical barrier, trapping AMPs and preventing them from reaching the epithelial surface where they exert their antimicrobial effects. This entrapment not only reduces the availability of functional AMPs but also contributes to the persistence of infections and the chronic inflammatory state characteristic of CF.
Implications of Dysregulated AMP Expression in CF
The dysregulation of AMP expression and function in CF has significant implications for disease progression and treatment. The reduced effectiveness of AMPs in CF patients leads to an increased susceptibility to infections, which are a major cause of morbidity and mortality in this population. Chronic infections drive the inflammatory response, leading to further lung damage and a decline in respiratory function over time.
Understanding the mechanisms behind AMP dysregulation in CF is critical for developing new therapeutic strategies aimed at enhancing mucosal defence. One approach that has been explored is the use of exogenous AMPs or AMP mimetics as potential therapies. These agents could be administered directly to the airways to supplement the deficient endogenous peptides and help control infections. Additionally, therapies aimed at correcting the ionic imbalance in CF airways, such as CFTR modulators, may also help restore the activity of AMPs and improve mucosal defence.
Potential Therapeutic Strategies Targeting AMPs in CF
Given the importance of AMPs in maintaining airway defence, several therapeutic strategies targeting these peptides are being explored for the treatment of CF. One approach involves the direct delivery of synthetic AMPs or AMP analogues to the lungs. These synthetic peptides are designed to mimic the structure and function of natural AMPs but are often engineered to be more stable and resistant to proteolytic degradation. By enhancing the antimicrobial activity in the airways, these therapies could help reduce bacterial load and inflammation in CF patients.
Another strategy focuses on modulating the expression of endogenous AMPs. For instance, certain cytokines, like IL-17 and IL-22, have been shown to upregulate AMP production in epithelial cells. Therapies that boost the levels of these cytokines or mimic their effects could enhance the natural production of AMPs in the airway mucosa. Additionally, CFTR modulators, which correct the underlying defect in CFTR function, may also indirectly enhance AMP activity by normalising the ionic environment in the airways.
Gene therapy is another potential avenue for restoring AMP function in CF. By delivering genes encoding AMPs or their regulators to the airway epithelium, it may be possible to increase the local production of these peptides and improve mucosal defence. Although still in the experimental stages, gene therapy holds promise as a long-term solution for managing infections in CF patients.
Conclusion
Antimicrobial peptides are essential components of the innate immune system, playing a critical role in protecting the airway mucosa from infection. In cystic fibrosis, however, the expression and function of these peptides are often compromised, leading to chronic infections and persistent inflammation. Understanding the mechanisms behind AMP dysregulation in CF is crucial for developing new therapeutic strategies aimed at enhancing mucosal defence. Approaches such as the delivery of synthetic AMPs, modulation of AMP expression, and gene therapy offer promising avenues for improving the health and quality of life for individuals with CF. As research in this field continues to advance, these strategies may one day provide effective solutions for managing the chronic infections that plague CF patients.
Journal Article: Geitani, R., et al., 2020. Expression and Roles of Antimicrobial Peptides in Innate Defense of Airway Mucosa: Potential Implication in Cystic Fibrosis. Frontiers in Immunology.
Summary by Faith Oluwamakinde
References
- Stolzenberg ED, Anderson GM, Ackerman MR, Huttner KM, Zasloff M. Epithelial antibiotic induced in states of disease. Proc Natl Acad Sci U S A. 1997;94(14):6976-6981.
- Bals R, Wang X, Meegalla RL, Wattler S, Weiner DJ, Nehls MC, Wilson JM. Mouse beta-defensin 3 is an inducible antimicrobial peptide expressed in the epithelia of multiple organs. Infect Immun. 1999;67(7):3542-3547.
- Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88(4):553-560.
- McCray PB Jr, Bentley L. Human airway epithelia express a beta-defensin. Am J Respir Cell Mol Biol. 1997;16(3):343-349.
- Murakami M, Ohtake T, Dorschner RA, Schittek B, Gallo RL. Cathelicidin antimicrobial peptide expression in sweat, an innate defence system for the skin. J Invest Dermatol. 2002;119(5):1090-1095.



