





- Patient presentation
- History
- Examination
- Investigations
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & answers
- MCQs
Patient Presentation
Hubert, a 67 year-old retired male, diagnosed with generalized myasthenia gravis positive for anti-acetylcholine receptor autoantibodies, was treated with efgartigimod for disease exacerbation.
Acknowledgement
This case study was provided by Prof. Olivier Boyer (M.D., Ph.D., Head of the Department of Immunology, Rouen University Hospital, France) and Dr. Henri Gondé (Pharm.D., Ph.D., Associate Professor of Pharmaceutical Technology) of the Faculty of Medicine and Pharmacy of Rouen, Normandy University, France. The authors would like to thank Dr Marie-Agnès Dragon-Durey (M.D., Ph.D., Department of Immunology, Georges Pompidou University Hospital, AP-HP, France), Prof. Jan De Bleecker (M.D., Ph.D., Department of Neurology, Ghent University Hospital, Belgium) and Dr. Joost Raaphorst (M.D., Ph.D., Department of Neurology, Amsterdam University Medical Center, Netherlands) for their critical reading of this case study.
History
The symptoms began two years ago with intermittent diplopia and ptosis, resulting in the diagnosis of myasthenia gravis positive for anti-acetylcholine receptor (AChR) antibodies and initiation of treatment with the acetylcholinesterase inhibitor pyridostigmine. However, a few months later, symptoms worsened, including complete oculomotor paresis in both eyes, swallowing difficulties, and generalized fatigability, indicating generalized myasthenia gravis. Azathioprine and intravenous immunoglobulins were introduced, but neither treatments sufficiently improved the clinical condition. Subsequently, treatment with glucocorticoids was initiated, resulting in a slight improvement in the myasthenia gravis symptoms. Unfortunately, the symptoms worsened when the glucocorticoid dosage was reduced. Due to the limited efficacy of the previous treatments, the patient was treated with efgartigimod.
- Past medical history:
Type 2 diabetes for 15 years, managed with sitagliptin/metformin, gliclazide and insulin glargine. - Surgical history:
Surgeries for a hip fracture, left elbow dislocation, and left radial head fracture resulting from a ladder fall 19 years ago. - Family history
His sister has Type 2A von Willebrand disease. - Travel history
None - Social history
Divorced
Living alone
No children - Medication:
Pyridostigmine 420 mg/day
Azathioprine 200 mg/day
Prednisone initiated at 80 mg/day and reduced to 20 mg/day
Intravenous immunoglobulins every 6 weeks
Examination
General
- Hubert looks tired
- No pallor
- No cutaneous signs
- No cough
Vitals
- Heart rate: 82/min
- Blood pressure: 125/80 mmHg
- Temperature: 36.9°C
- Oxygen saturation: 98%
Cardiovascular
- Normal heart sounds
- All pulses present
Respiratory
- No dyspnoea (laboured breathing)
- No chest deformity
- No crackling sound on the lungs or wheezing
Abdomen
- Normal
Neurological
- Bilateral ptosis (drooping eyelids)
- Diplopia
- Swallowing difficulties
- Normal pupillary reflexes
- Normal reflexes on upper and lower limbs
- Normal cognition
Musculoskeletal system
- Motor fatigability of the lower and upper limbs worsened with activity
- Unable to ride a bicycle for more than 15 minutes
Investigations
| Examination | Value | Normal limits |
|---|---|---|
| RBC | 4.60 | 4.5-5.8 x109/L |
| HB | 14.1 | 13-17.5 g/L |
| WBC | 5.8 | 4-10 x109/L |
| Neutrophils | 4.73 | 1.7-7.5 x109/L |
| Eosinophils | 0.02 | 9/L |
| Basophils | 0.03 | 9/L |
| Lymphocytes | 1.2 | 1.2-4 x109/L |
| Monocytes | 0.30 | 0.2-1 x109/L |
| Platelets | 229 | 150-400 x109/L |
| Alkaline phosphatase | 46 | 40-130 IU/L |
| GGT | 12 | 10-55 IU/L |
| ALT | 22 | 10-50 IU/L |
| ALP | 26 | 10-50 IU |
| Blood creatinine | 67 | 59-104 µmol/L |
| Blood glucose | 2.39 | 0.7-1.1 g/L |
| Na+ | 142 | 135-145 mmol/L |
| K+ | 4 | 3.6-4.6 mmol/L |
| Cl- | 103 | 96-106 µmol/L |
Before treatment with efgartigimod
| Total protein | 68 | 60-80 g/L |
|---|---|---|
| Albumin | 43 | 35-50 g/L |
| IgG | 12 | 8-16 g/L |
| Anti-AChR antibodies | 35.0 | |
| Anti-MuSK antibodies | Negative | |
| Antinuclear antibodies | Negative |
After a cycle of treatment with efgartigimod (4 weekly infusions)
| Total protein | 60 | 60-80 g/L |
|---|---|---|
| Albumin | 42 | 35-50 g/L |
| IgG | 5 | 8-16 g/L |
| Anti-AChR antibodies | 14 | |
| Anti-MuSK antibodies | Negative | |
| Antinuclear antibodies | Negative |
Discussion
Efgartigimod is indicated in combination with standard treatment for adult patients with generalized MG positive for anti-AChR autoantibodies. It is an immunomodulatory treatment that blocks the function of the neonatal Fc receptor (FcRn). Efgartigimod is administered at a dose of 10 mg/kg through intravenous infusion or 1000 mg via subcutaneous injection. A treatment cycle consists of one weekly administration for 4 weeks. Subsequent treatment cycles are administered based on clinical evaluation. Of note, some patients with refractory MG may require more frequent dosing to achieve a clinical response (1).
Myasthenia gravis
MG is a rare autoimmune disease affecting the neuromuscular junction, with a prevalence of 150-200 cases per million people and an incidence rate of 4-30 cases per million person-years. MG presents a bimodal distribution with two peaks of incidence. Early-onset MG predominantly affects women in the third decade of life, while late-onset MG primarily affects elderly males after the 6th decade, as is the case with Hubert, who was diagnosed at the age of 65 (2).
MG is characterized by muscle weakness that worsens with exertion. Ocular manifestations are common, with diplopia (double vision) and ptosis (drooping of the upper eyelid). Ocular MG is characterized by almost exclusively ocular symptoms and can evolve to generalized MG. In generalized MG, ocular symptoms are accompanied with muscle weakness in other areas, such as the limbs, axial muscles, and/or oropharyngeal muscles. For the clinical classification of MG established by the Myasthenia Gravis Foundation of America (MGFA), you can refer to the clinical case “My eyes cross at twilight“.
The muscle weakness in MG is caused by autoantibodies that impair neuromuscular transmission. In most patients, the antibodies are directed against AChR. Anti-AChR antibodies impair neuromuscular transmission at the post-synaptic junction by triggering complement-mediated lysis of the postsynaptic membrane, blocking AChR, thereby preventing the interaction of ACh with its receptor, or causing internalization of AChR, reducing their number on the postsynaptic membrane. Alternative autoantibodies can also cause MG, including anti-MuSK (muscle-specific kinase, a tyrosine kinase involved in the clustering of AChR at the neuromuscular junction) or anti-LRP4 (low-density lipoprotein receptor-related protein 4, the agrin receptor involved in MuSK activation). In addition, autoantibodies directed to intracellular striated muscle proteins may also be found, such as anti-titin antibodies, which are associated with thymoma in patients with MG (3). You can refer to the clinical case “My eyes cross at twilight” for more information on the pathophysiology of MG.
FcRn
FcRn is structurally related to MHC class I molecules. Both proteins are heterodimers composed of an α heavy chain, non-covalently linked to the β2-microglobulin chain. The α chain includes 3 extracellular domains (α1, α2, and α3), a transmembrane domain, and a short intracellular domain.
In contrast to MHC molecules, FcRn exhibits low polymorphism in the human population and is unable to present antigenic peptides to T cells. FcRn is broadly expressed throughout the body, particularly on the surface of vascular endothelial, hematopoietic (especially monocytes and macrophages), and epithelial cells (intestine, liver, kidneys, lungs, blood-brain barrier, mammary gland, and placenta) (4).
FcRn was initially identified in rodents as the receptor responsible for transporting maternal IgG across the intestinal epithelium of neonatal offspring via maternal milk, leading to its designation as the neonatal Fc receptor. In humans, FcRn facilitates the transcytosis of maternal IgG to the fetus through the placenta, mostly during the third trimester of pregnancy, and does not appear to play a major role in the postnatal transfer of maternal IgG to the new born.
One of the major functions of FcRn throughout life is the recycling of IgG and albumin, accounting for their long half-life (≈ 21 days for IgG1, IgG2, IgG4, and albumin). Indeed, IgG, albumin, and other proteins are continuously ingested through pinocytosis by different cells (Figure 2). The resulting endosomes then undergo acidification (pH 5.0-6.5), leading to the degradation of the ingested proteins and a short blood half-life. FcRn is able to interact with IgG and albumin in a pH-dependent manner, leading to their recycling to the blood and therefore longer half-life.
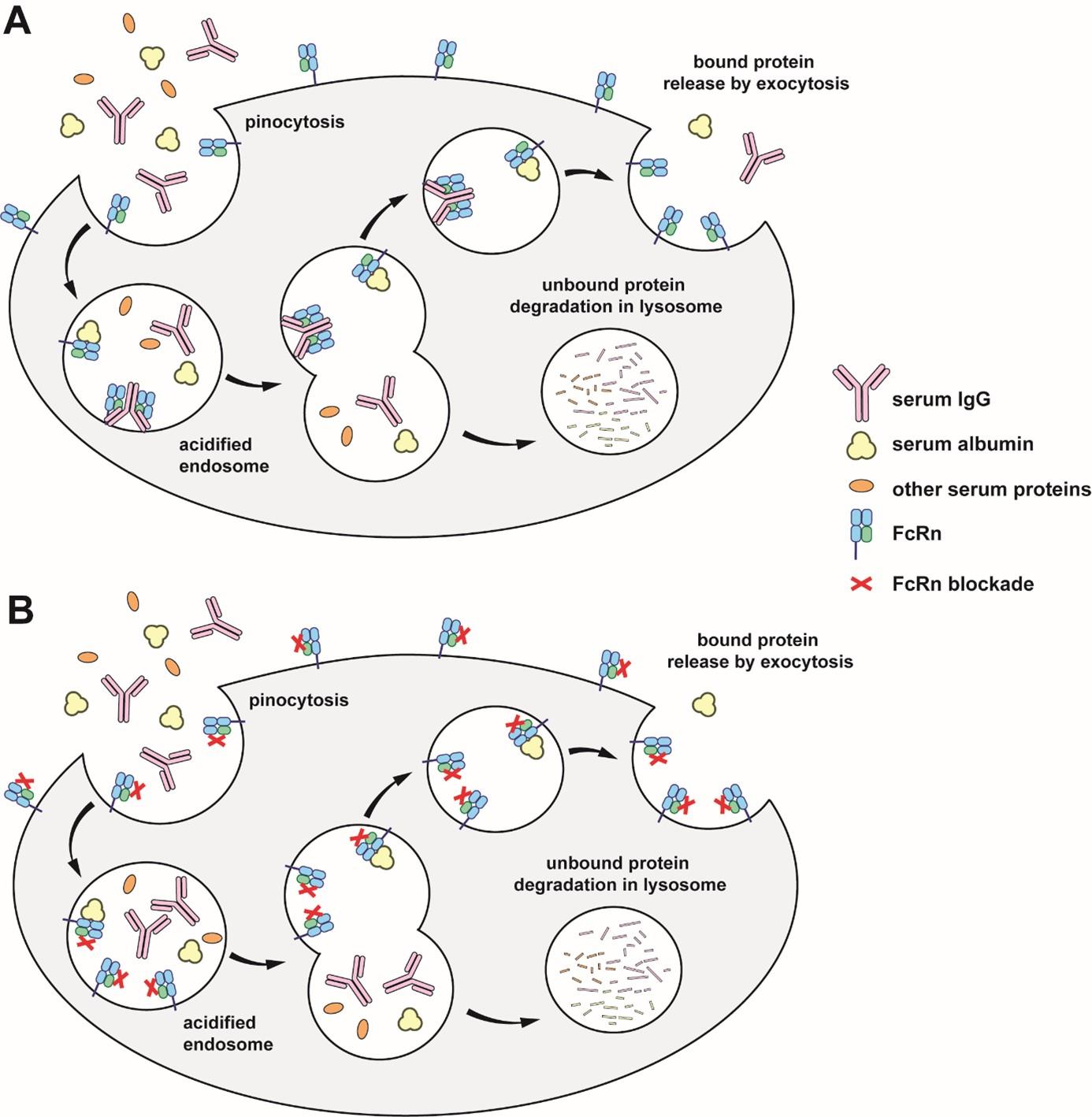
Figure 1: Recycling of endogenous IgG and albumin by FcRn and mode of action of FcRn blockers
(A) IgG, albumin, and other serum proteins are internalized through pinocytosis. Acidification of the endosome leads to binding of IgG and albumin to FcRn, but not to other proteins. IgG and albumin bound to FcRn are released by exocytosis, whereas proteins that are not bound to FcRn are degraded in lysosomes. This phenomenon takes place notably in macrophages and endothelial cells.
(B) FcRn blockers bind to FcRn at both neutral and acidic pH. Endogenous IgG are unable to interact with FcRn and are degraded in lysosomes rather than being released into the extracellular environment.
The binding of FcRn by IgG is mainly mediated by histidine residues (H310 and H435) located in the vicinity of CH2-CH3 interface of IgG (5). Histidine is the only amino acid whose protonation state changes near physiological pH values (pKa ≈ 6-7). At neutral pH, the aromatic imidazole ring of the side chain is neutral (non-protonated), whereas in an acidic environment (pH below 6), it becomes protonated (Figure 2).
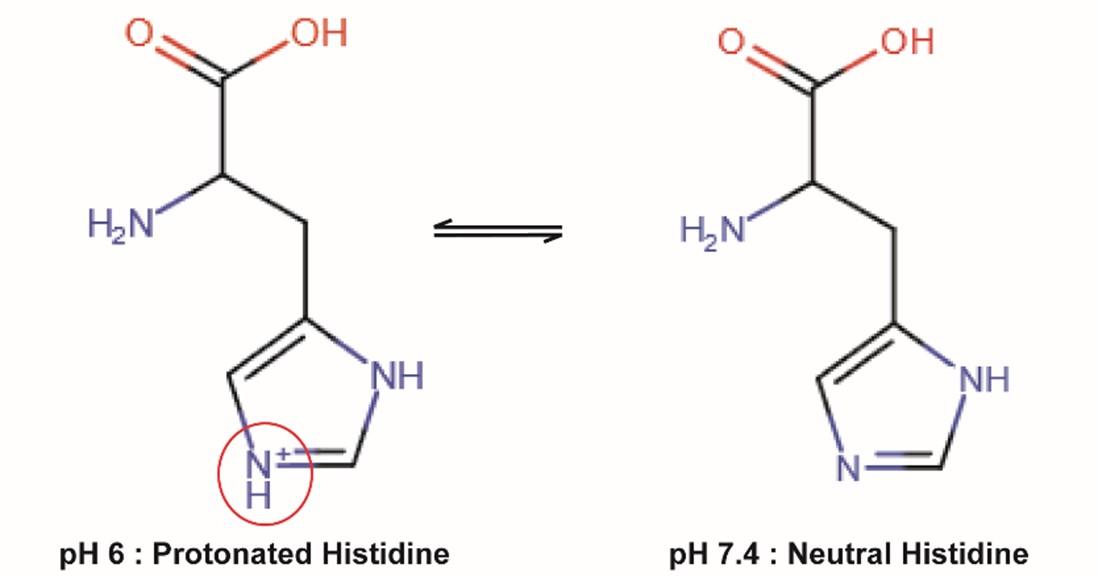
Figure 2: Protonation of Histidine according to pH values
The protonated form of histidine interacts with negatively charged residues. In the case of IgG, the protonated histidine residues interact with negatively charged residues located in the α2 and β2m domains of FcRn in the acidic environment of endosomes. Of note, the substitution of one of the histidine residues involved in the interaction with FcRn (H435 to R435) reduces the affinity for FcRn and accounts for the reduced half-life of IgG3 (9 days) compared to other subclasses that compete for FcRn binding (6). IgG molecules are heterotetramers (consisting of two identical heavy chains and two identical light chains) and thus have two sites of interaction with FcRn. Consequently, two FcRn molecules can bind to a single IgG molecule.
In contrast to IgG, the engagement of FcRn by albumin occurs in a 1:1 ratio, at a site distinct from that of IgG. Thus, FcRn can bind simultaneously one molecule of IgG and one molecule of albumin. Albumin does not directly interact with FcRn through histidine residues. Instead, it interacts with a loop located in the α2 domain of FcRn that is stabilized by a histidine residue (H166) of the α1 domain in a pH-dependent manner (7). Similarly to IgG, albumin exhibits very low affinity for FcRn at neutral pH but high affinity at the acidic pH of endosomes.
IgG and albumin bound to FcRn are recycled, i.e. transported back to the cell surface. Upon fusion with the plasma membrane and pH rising, IgG and albumin dissociate from FcRn and are released back to the extracellular environment. Other proteins, including excess IgG and albumin not bound to FcRn, are directed to the lysosomal pathway leading to their degradation.
FcRn blockade
Blocking the FcRn prevents its interaction with endogenous IgG, including pathogenic autoantibodies. As a result, IgGs are no longer recycled and are degraded more rapidly (Figure 2B). This is beneficial in conditions where pathogenic autoantibodies play a critical role, such as in MG. The blockade of FcRn is achieved either through engineered forms of the Fc fragment of IgG (e.g., efgartigimod) or by using monoclonal antibodies that target FcRn (e.g., rozanolixizumab and nipocalimab) (Figure 3).
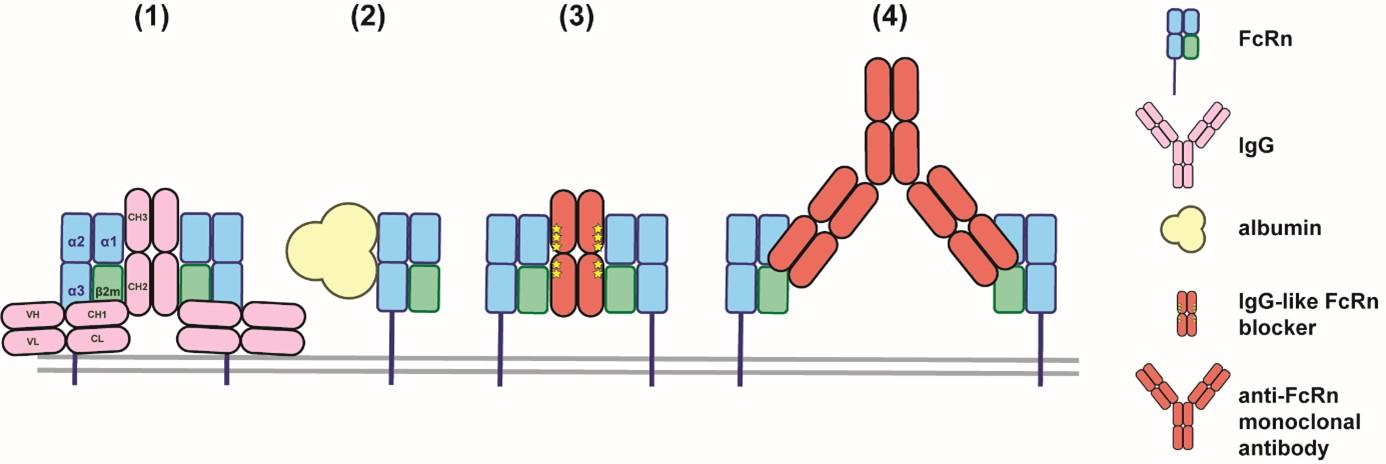
Figure 3: Interaction of endogenous proteins and biologics with FcRn
FcRn is involved in the recycling of IgG (1) and albumin (2) by interacting at different sites of the receptor in a pH-dependent manner. FcRn blockade can be achieved with IgG-like molecules that interact with FcRn through the Fc part (e.g., efgartigimod) (3) or with monoclonal antibodies targeting FcRn and blocking the interaction between IgG and FcRn (e.g., rozanolixizumab and nipocalimab) (4).
Efgartigimod was approved in 2021 in USA and 2022 in Europe for the treatment of generalized MG in adults with anti-AChR antibodies. In Japan, efgartigimod is approved for generalized MG patients regardless of antibody status. Efgartigimod is an engineered human IgG1 antibody Fc-fragment that contains five point mutations (MST-HN: M252Y, S254T, T256E, H433K, N434F). Compared to wild-type IgG, these mutations confer to Efgartigimod a higher affinity to Fc region for FcRn at both pH 6 and pH 7.2. As a result, the binding sites for IgG on FcRn are saturated, preventing the binding of endogenous IgG without affecting serum albumin. In the pivotal Phase III trial, one cycle of treatment with efgartigimod resulted in a mean maximum reduction of 57.6% in anti-AChR antibodies and 61.3% in total IgG, with no reduction in albumin levels (Figure 4) (8). The transient reduction in IgG levels by efgartigimod may increase the risk of infections, particularly upper respiratory tract and urinary tract infections. The administration of live or live-attenuated vaccines is not recommended for patients undergoing treatment with efgartigimod. All other vaccines should be administered at least 2 weeks after the last infusion of a treatment cycle or 4 weeks before the start of a next cycle. Efgartigimod reduces plasma concentrations of molecules that bind to the FcRn, including intravenous immunoglobulins, therapeutic monoclonal antibodies and antibody derivatives containing the Fc domain of the IgG subclass. Therefore, it is recommended to postpone the initiation of treatment with these biologics until 2 weeks after the last dose of an efgartigimod treatment cycle. In cases where treatment with biologics is already ongoing at the start of efgartigimod, the therapeutic response to these agents should be closely monitored. Plasma exchange and plasmapheresis may reduce circulating levels of efgartigimod.
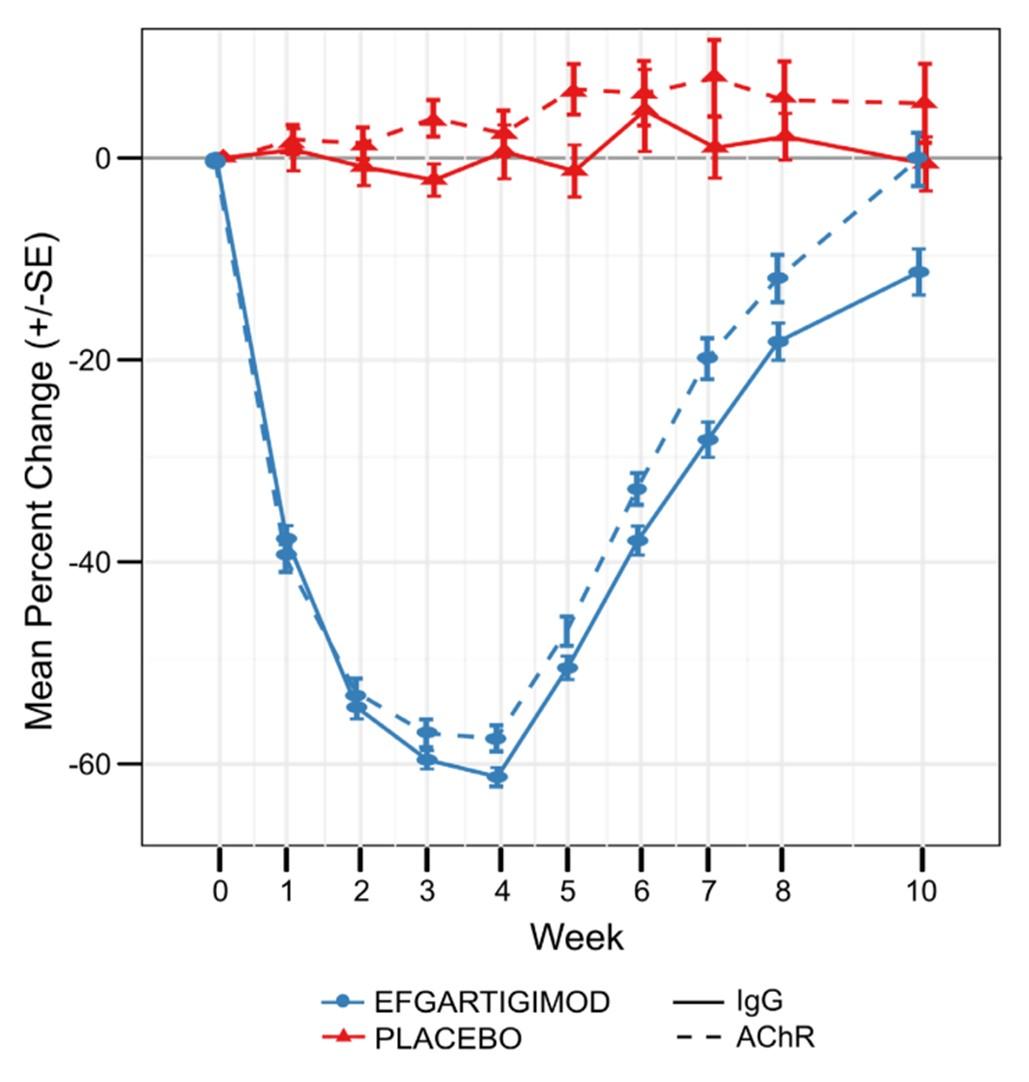
Figure 4: Mean change in antibody levels after one cycle of efgartigimod treatment (4 weekly infusions).
This figure is extracted from the supplementary appendix of the Phase III trial publication (8). The trial enrolled 167 patients with generalized myasthenia gravis (MG), including 129 patients positive for anti-AChR antibodies (65 in the efgartigimod group and 64 in the placebo group).
Rozanolixizumab and nipocalimab are two monoclonal antibodies that selectively bind and block the IgG binding site on human FcRn while not overlapping with albumin binding site.
Rozanolixizumab is a humanized IgG4 monoclonal antibody approved for treatment of generalized MG in adult patients positive for anti-AChR or anti-MuSK antibodies. Rozanolixizumab is administered subcutaneously once a week for 6 weeks, with the dose adjusted for patient bodyweight. In the pivotal phase III trial, rozanolixizumab 7 mg/kg and 10 mg/kg achieved a median maximum reduction in AChR antibodies of respectively 73% and 82%, respectively. Similarly, the maximum reduction in total IgG was 73% and 79% for rozanolixizumab 7 mg/kg and 10 mg/kg (9)
Nipocalimab is a fully human IgG1 aglycosylated monoclonal. The absence of ubiquitous N-glycans at N197 precludes effector functions by abolishing Fc binding to Fc γ receptors (FcγR) and the C1q fraction of the complement, while preserving the binding to FcRn. Nipocalimab is currently being tested in a phase III clinical trial for generalized MG (NCT04951622).
Treatment
The aim of MG treatments is to achieve MGFA minimal manifestations or no symptoms or functional limitations (10,11).
Conventional treatments
Conventional treatments for MG include symptomatic and immunosuppressive therapies.
- Symptomatic treatments: cholinesterase inhibitors (g., pyridostigmine) slow down the hydrolysis of acetylcholine in the synaptic cleft. They are included in the initial treatment of MG. The dose should be adjusted based on symptoms and side effects.
- Immunosuppressive therapies: Immunosuppressive therapies are indicated when therapeutic goals are not achieved with cholinesterase inhibitors alone. Immunosuppressive treatments encompass glucocorticoids and non-steroidal immunosuppressants. Non-steroidal immunosuppressive agents that can be used in MG include azathioprine, cyclosporine, mycophenolate mofetil, methotrexate, and tacrolimus. Non-steroidal immunosuppressive agents should be added to corticosteroids in cases of significant side effects, insufficient treatment response, or when the corticosteroid dose cannot be reduced due to relapses.
Adjunct therapies
- Plasma exchange and intravenous immunoglobulins can be used as short-term treatments for patients with refractory generalized MG with life-threatening symptoms, such as respiratory deficiency or dysphagia.
- Resection of the thymus (thymectomy) is required if thymoma or thymic enlargement are detected by imaging. Thymectomy should be considered early in the disease for non-thymomatous, generalized MG patients with AChR antibodies, aged 18 to 50 years (12).
Biologics:
In addition to FcRn blockers, several biologics are currently used in the treatment of refractory MG.
- Rituximab is a chimeric monoclonal antibody targeting the CD20 protein expressed on the surface of B cells (except for progenitor B cells and mature plasma cells, which do not express it). Binding of rituximab to CD20-expressing cells results in their depletion by triggering antibody-dependent cellular cytotoxicity through the activation of NK cells, antibody-dependent cellular phagocytosis by activated macrophages, and complement-dependent cytotoxicity related to the activation of the classical complement pathway leading to membrane attack complex formation. The efficacy of rituximab in refractory AChR-positive MG is uncertain, whereas it should be considered as an early therapeutic option in patients with anti-MuSK autoantibodies.
- Eculizumab is a humanized monoclonal antibody targeting the C5 component of the complement system. Binding of eculizumab to C5 inhibits the terminal pathway of complement activation, preventing the formation of the membrane attack complex and reducing the damage caused by complement activation triggered by autoantibodies in MG. Eculizumab is currently approved for AChR positive refractory generalized MG.
- Ravulizumab is a humanized monoclonal antibody derived from eculizumab that was engineered to extend its duration of action. This was accomplished by introducing four point mutations, including two in the Fc region and two in the complementarity-determining regions (CDRs) of the variable region, the latter reducing target-mediated drug disposition, an elimination pathway of therapeutic antibodies associated with high-affinity binding to the target, promoting their elimination (13). Therefore, ravulizumab is administered every 8 weeks
- Zilucoplan is a macrocyclic peptide that binds to C5 and inhibits its cleavage, thereby preventing the formation of membrane attack complex. Zilucoplan is administered subcutaneously daily and is approved for the treatment of adults with generalized MG.
Final Outcome
After a cycle of treatment with efgartigimod (4 weekly infusions), Hubert experienced a 58% decrease of AChR antibody levels and sustained clinical improvement, which led to the discontinuation of corticosteroids and intravenous immunoglobulins.
References
- Remijn-Nelissen L, Tannemaat MR, Ruiter AM, Campman YJM, Verschuuren JJGM. Efgartigimod in refractory autoimmune myasthenia gravis. Muscle Nerve. 2024;70(3):325‑32.
- Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. J Clin Med. 2021;10(11):2235.
- Moura J, Sousa AP, Samões R, Carneiro P, Neves E, Silva AM, et al. Anti-titin antibodies in a cohort of myasthenia gravis patients. J Thorac Dis. 2024;16(2):973‑8.
- Pyzik M, Kozicky LK, Gandhi AK, Blumberg RS. The therapeutic age of the neonatal Fc receptor. Nat Rev Immunol. 2023;23(7):415‑32.
- Martin WL, West AP, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7(4):867‑77.
- Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun. 2011;2:599.
- Sand KMK, Dalhus B, Christianson GJ, Bern M, Foss S, Cameron J, et al. Dissection of the neonatal Fc receptor (FcRn)-albumin interface using mutagenesis and anti-FcRn albumin-blocking antibodies. J Biol Chem. 2014;289(24):17228‑39.
- Howard JF, Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526‑36.
- Bril V, Benatar M, Andersen H, Vissing J, Brock M, Greve B, et al. Efficacy and Safety of Rozanolixizumab in Moderate to Severe Generalized Myasthenia Gravis: A Phase 2 Randomized Control Trial. Neurology. 2021;96(6):e853‑65.
- Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. 2016;87(4):419‑25.
- Narayanaswami P, Sanders DB, Wolfe G, Benatar M, Cea G, Evoli A, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021;96(3):114‑22.
- Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med. 2016;375(6):511‑22.
- Sheridan D, Yu ZX, Zhang Y, Patel R, Sun F, Lasaro MA, et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. PloS One. 2018;13(4):e0195909.
Evaluation – Questions & answers
Why blocking FcRn can be beneficial in the treatment of myasthenia gravis?
Myasthenia gravis is caused by pathogenic autoantibodies that disrupt the neuromuscular transmission. Blocking FcRn accelerates the clearance of endogenous IgG from the bloodstream, including the pathogenic autoantibodies. This improves neuromuscular transmission and alleviates clinical symptoms.
What are the strategies to block FcRn for therapeutic applications?
FcRn blockade can be achieved using IgG-like biologics with higher affinity for FcRn at both neutral and acidic pH (e.g., efgartigimod) or with monoclonal antibodies targeting FcRn (e.g., rozanolixizumab and nipocalimab).
Why are histidine key residues for IgG to interact with FcRn?
The imidazole group of histidine is non-protonated at neutral pH but protonated (positively charged) at acidic pH. Protonation of histidine residues in the Fc fragment of IgG promotes their interaction with negatively charged residues of FcRn within the acidic environment of endosomes and their subsequent release at neutral pH.
Which proteins are protected from lysosomal degradation by FcRn?
- Endogenous IgG
- Exogenous IgG (monoclonal antibodies)
- Albumin
What are the biologics that can be used in the treatment of generalized myasthenia gravis?
- Efgartigimod, an engineered Fc fragment with a higher affinity for FcRn than wild-type IgG at both pH 6 and pH 7.2.
- Rozanolixizumab and nipocalimab, monoclonal antibodies directed to FcRn
- Rituximab: a B cell-depleting monoclonal antibody directed to CD20
- Eculizumab and ravulizumab: monoclonal antibodies directed to C5 fraction of complement
- Zilucoplan: a macrocyclic peptide that binds to C5 fraction of complement



