





- Patient Presentation
- History
- Differential Diagnosis
- Examination
- Investigation
- Diagnosis
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & answers
- MCQ
Patient Presentation
A 10-month-old boy presented to a private maternal and child health hospital with severe irritability and floppiness. He had been fully immunized and was breastfed until 9 months of age.
History
Apart from a history of recurrent respiratory tract infections, his past medical history was unremarkable. At presentation a diagnosis of meningitis or meningoencephalitis was considered.
Differential Diagnosis
- Viral meningo-encephalitis
- Bacterial meningitis
- Herpes Simplex encephalitis
- Lower respiratory tract infection
Examination
On Admission:
General examination:
- Irritable and febrile with temperature 38.5oC
- Slightly under weight for age at 7 Kg
Central nervous system:
- Floppy, but moving all limbs. No other abnormalities detected
Respiratory system:
- Crackles in chest bilaterally
No other findings of note
Investigation
Routine investigations on admission showed a raised white cell count, with a neutrophil predominance:
Hb 13.5 g/dL
- platelets 523 x 109/L
- WCC 21.65 x 109/L
- Neuts 12.67 (1-5)
- Lymphs 7.45 (4-12)
- Mono 1 (0.2-1.2)
- Eos 0.02 (0-1)
- Baso 0.15 (0-0.1)
Chemistry
- Normal
Respiratory
- Virus screen (21.09.11) sputum: negative
CSF specimens were taken on admission and then 3 days later. The profile was consistent with an uncomplicated aseptic meningitis. Enterovirus PCR was positive on the second CSF sample.
| CSF | Date 1 | Date 2 (3d later) |
|---|---|---|
| Colour | Turbid | Turbid |
| Supernatant | Clear | Clear |
| Total white cells (0-5 cells/uL) | 50 | 44 |
| Polymorphs (0-5 cells/uL) | 35 | 15 |
| Monos (0-5 cells/uL) | 15 | 29 |
| Red cells (0-5) | 0 | 2 |
| Total protein (0.15-0.45g/L) | 0.46 | 0.26 |
| Glucose (3.3-4.4mmol/L) | 3.8 | 3.5 |
| IgG (4.8-58.6mg/L) | ||
| EV PCR | Negative | Positive (Ct 28) |
| HSV PCR | Negative |
The enteroviral infection and history of recurrent bacterial infections raised the suspicion of a primary immunodeficiency and screening investigations were undertaken.
Measurement of various classes of immunoglobulins showed a virtual absence of IgA, IgM and IgG subclasses1-4.
| Immunoglobulin levels (g/L) | Vaccine responses | ||
|---|---|---|---|
| IgA | Tetanus Ab | 0.1IU/mL] | |
| IgM | Diphtheria Ab | 0.1IU/mL] | |
| IgG | 0.1 [3.5-11.8] | H influenzae Ab | 0.84 mg/L] |
| IgG1 | S pneumoniae IgG | 9.2mg/L] | |
| IgG2 | |||
| IgG3 | |||
| IgG4 |
In addition, vaccine responses to tetanus, diphtheria, Haemophilis influenzae and Streptococcus pneumonia were found to be completely absent. This picture was highly suggestive of a primary immunodeficiency and flow cytometric lymphocyte immunophenotyping was requested.
Total T lymphocyte and CD4 and CD8 T lymphocyte subsets were normal. But B lymphocytes were absent.
| Reference range | 27.09.2011 | |
|---|---|---|
| CD45 cells | 4000-12000 cells/uL | 1932 |
| Total T-cells Abs | 1900-5900 cells/uL | 1769 |
| Total T cells % | 49-76% | 92 |
| CD4 cells Abs | 1400-4300 cells/uL | 1266 |
| CD4 cells % | 31-56% | 65 |
| CD8 cells Abs | 500-1700 cells/uL | 511 |
| CD8 cells % | 12-24% | 26 |
| Total B cells Abs | 610-2600 cells/uL | 1 |
| Total B cells % | 14-37% | 0 |
| Total NK cells Abs | 160-950 cells/uL | 138 |
| Total NK cells % | 3-15% | 7 |
In addition, stool from the child was also enterovirus positive. Sequencing of the PCR product was performed to type the enterovirus and it was identified as polio type 3 virus.
Stool specimens were analyzed further to determine whether this was vaccine or wild type virus. The virus was confirmed to be a vaccine-derived poliovirus with 12 nucleotide substitutions in the VP1 gene.
Diagnosis
A presumptive diagnosis of X-linked agammaglobulinaemia was made, complicated by Poliovaccine-derived paralytic poliomyelitis.
Discussion
To understand how this child succumbed to vaccine derived paralytic poliomyelitis, it is first necessary to understand the life cycle of Poliovirus and how the vaccine works. We will then describe how X-linked agammaglobulinaemia was the underlying aetiology in this child.
Lets first look at how wild-type poliovirus and vaccine strain replication occurs
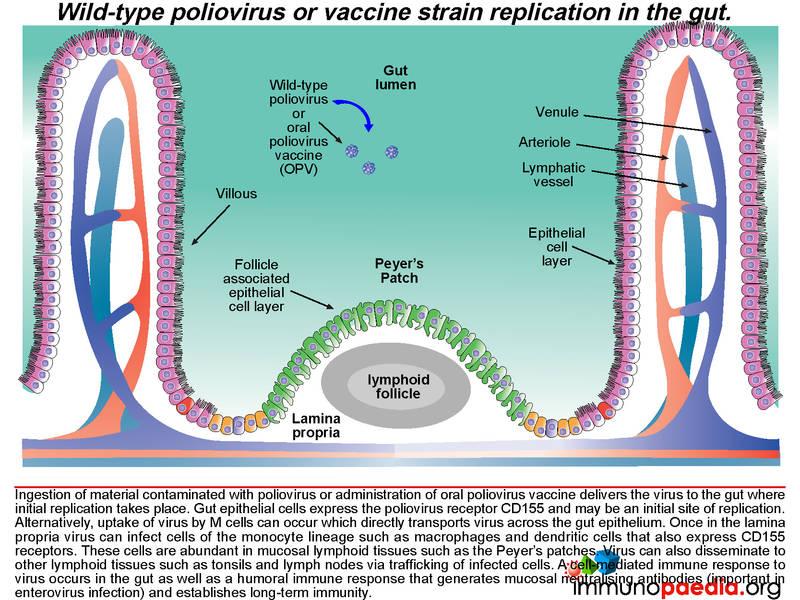
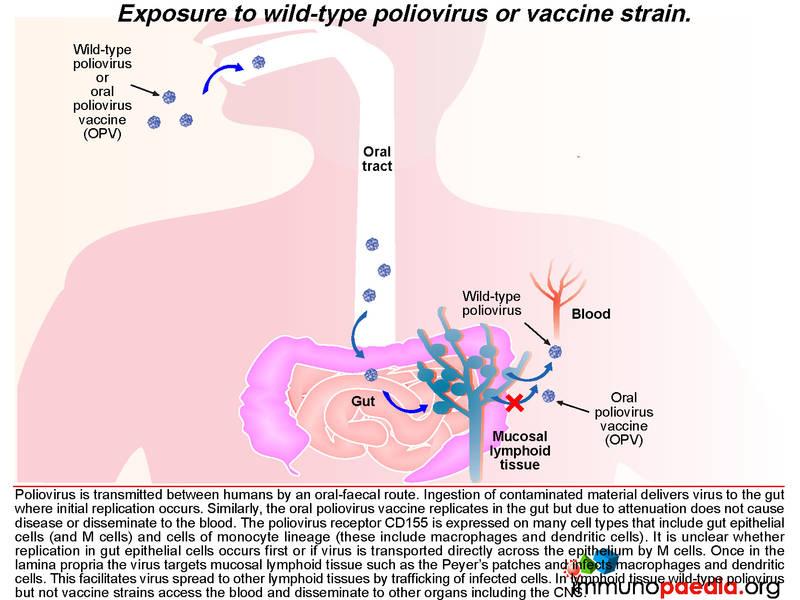 Poliovirus transmission between humans is by an oral-faecal route. Ingestion of contaminated material delivers virus to the gut where initial replication occurs. Gut epithelial cells express the poliovirus receptor CD155 and may be an initial site of replication. Alternatively, uptake of virus by M cells can occur which directly transports virus across the gut epithelium.
Poliovirus transmission between humans is by an oral-faecal route. Ingestion of contaminated material delivers virus to the gut where initial replication occurs. Gut epithelial cells express the poliovirus receptor CD155 and may be an initial site of replication. Alternatively, uptake of virus by M cells can occur which directly transports virus across the gut epithelium.
Once in the lamina propria virus can infect cells of the monocyte lineage such as macrophages and dendritic cells that also express CD155 receptors. These cells are abundant in mucosal lymphoid tissues such as the Peyer’s patches. Virus can also disseminate to other lymphoid tissues such as tonsils and lymph nodes via trafficking of infected cells. A cell-mediated immune response to virus occurs in the gut as well as a humoral immune response that generates neutralising antibodies which is of particular importance in enterovirus infection and also in establishing long-term immunity.
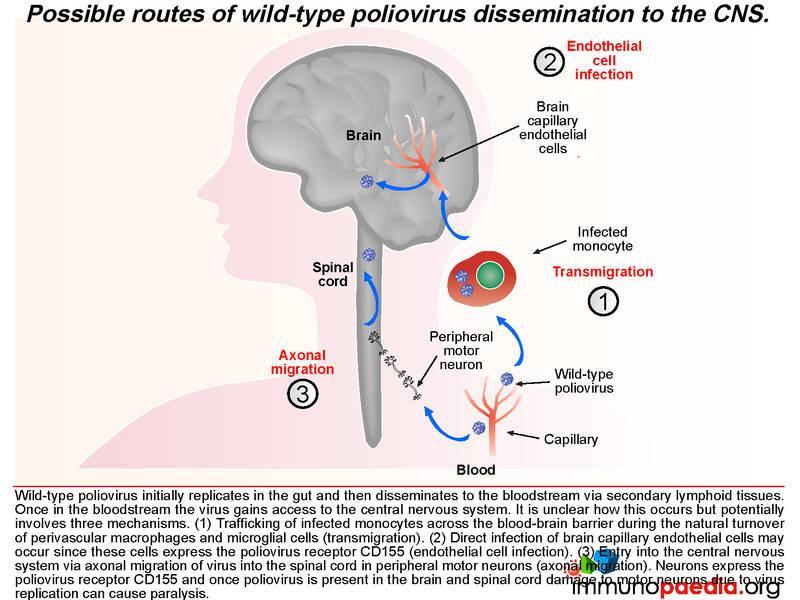
 Once in lymphoid tissue, wild-type poliovirus can enter the blood and spread to other organs such as the brain. It is unclear how the virus accesses the central nervous systembut this could involve three possible routes: 1) by infected monocytes that can carry the virus across the blood-brain barrier during maturation into perivascular macrophages and microglial cells; 2) poliovirus could infect brain capillary endothelial cells since they express CD155; 3) poliovirus is neurotropic and could access the brain via axonal transfer from peripheral motor neurons into the spinal cord. Neurons express CD155 and once poliovirus is present in the brain or spinal cord it damages motor neurons leading to paralysis. Typically only 1% of infected individuals develop symptoms of CNS infection.
Once in lymphoid tissue, wild-type poliovirus can enter the blood and spread to other organs such as the brain. It is unclear how the virus accesses the central nervous systembut this could involve three possible routes: 1) by infected monocytes that can carry the virus across the blood-brain barrier during maturation into perivascular macrophages and microglial cells; 2) poliovirus could infect brain capillary endothelial cells since they express CD155; 3) poliovirus is neurotropic and could access the brain via axonal transfer from peripheral motor neurons into the spinal cord. Neurons express CD155 and once poliovirus is present in the brain or spinal cord it damages motor neurons leading to paralysis. Typically only 1% of infected individuals develop symptoms of CNS infection.
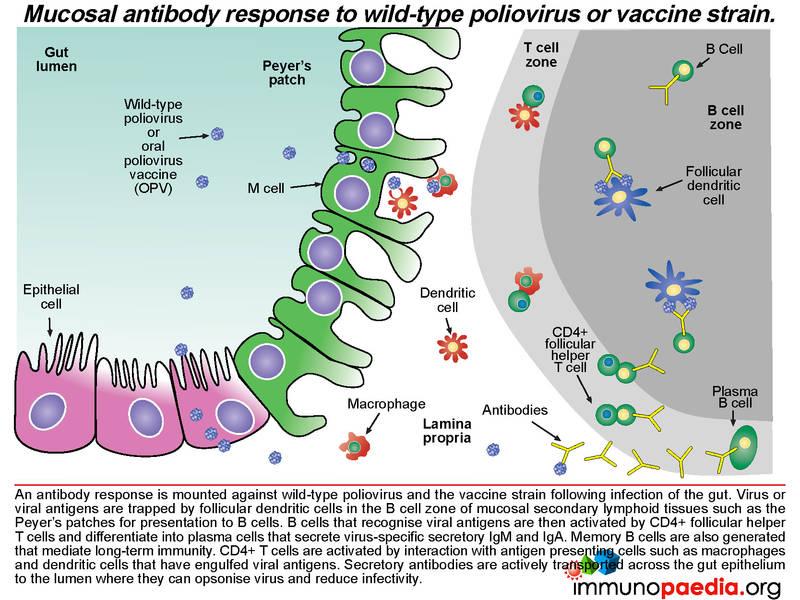
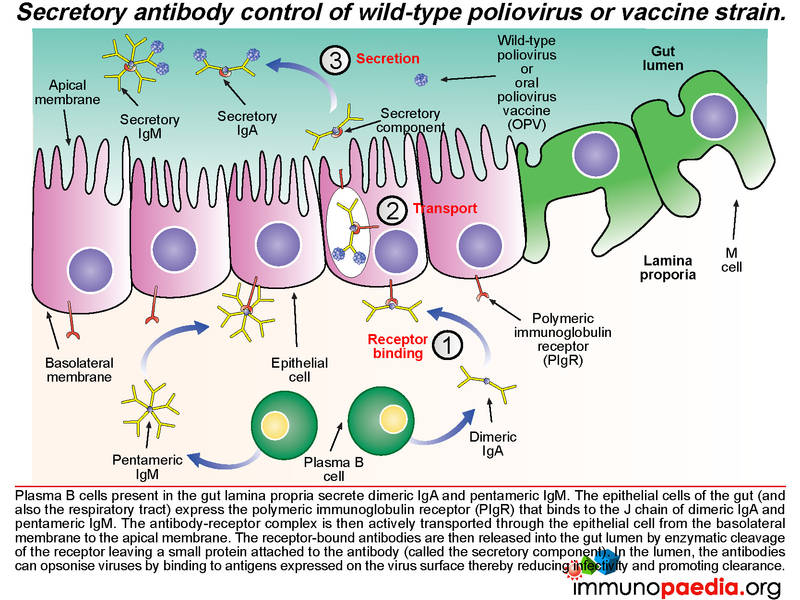
 An antibody response is mounted against poliovirus following infection of the gut. Whole virus or viral antigens are trapped by follicular dendritic cells in the B cell zone of the mucosal secondary lymphoid tissues such as the Peyer’s patches. B cells that recognise viral antigens are activated by CD4+ follicular helper T cells and differentiate into plasma cells that produce secretory IgM and IgA. CD4+ T helper cells are activated by interaction with antigen presenting cells such as macrophages and dendritic cells that have engulfed viral antigens. B cells present in the gut lamina propria secrete dimeric IgA and pentameric IgM. The epithelial cells of the gut express the polymeric immunoglobulin receptor (PIgR) that binds to the J chain of dimeric IgA and pentameric IgM.
An antibody response is mounted against poliovirus following infection of the gut. Whole virus or viral antigens are trapped by follicular dendritic cells in the B cell zone of the mucosal secondary lymphoid tissues such as the Peyer’s patches. B cells that recognise viral antigens are activated by CD4+ follicular helper T cells and differentiate into plasma cells that produce secretory IgM and IgA. CD4+ T helper cells are activated by interaction with antigen presenting cells such as macrophages and dendritic cells that have engulfed viral antigens. B cells present in the gut lamina propria secrete dimeric IgA and pentameric IgM. The epithelial cells of the gut express the polymeric immunoglobulin receptor (PIgR) that binds to the J chain of dimeric IgA and pentameric IgM.
 The antibody-receptor complex is then transported through the epithelial cells from the basolateral membrane to the apical membrane and released into the gut lumen. This is achieved by enzymatic cleavage of the receptor, which leaves a small protein attached to the antibody (called the secretory component). These antibodies can opsonise polioviruses in the lumen by binding to antigens expressed on the surface of the virion and thereby reduce infectivity and promote clearance.
The antibody-receptor complex is then transported through the epithelial cells from the basolateral membrane to the apical membrane and released into the gut lumen. This is achieved by enzymatic cleavage of the receptor, which leaves a small protein attached to the antibody (called the secretory component). These antibodies can opsonise polioviruses in the lumen by binding to antigens expressed on the surface of the virion and thereby reduce infectivity and promote clearance.
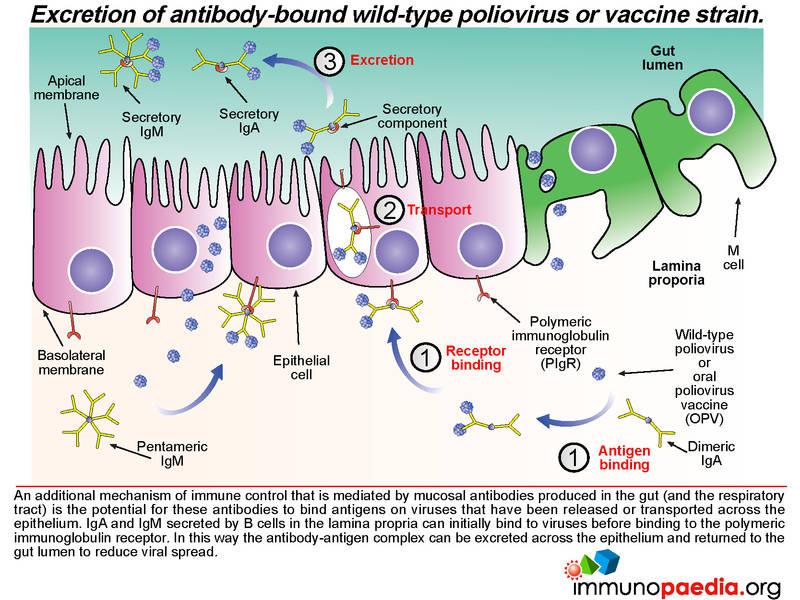
 Not only does mucosal antibody neutralize virus in the gut lumen, it can also clear virus on the basolateral side of the epithelium. Antibody-bound virus can be transported back across the epithelial cell layer into the gut lumen by binding to the polymeric immunoglobulin receptor expressed on the basolateral surface of the epithelial cell.
Not only does mucosal antibody neutralize virus in the gut lumen, it can also clear virus on the basolateral side of the epithelium. Antibody-bound virus can be transported back across the epithelial cell layer into the gut lumen by binding to the polymeric immunoglobulin receptor expressed on the basolateral surface of the epithelial cell.
How does the oral poliovirus vaccine (OPV) work?
There are 3 serotypes of wild poliovirus (poliovirus 1, 2 and 3) and OPV contains attenuated strains of these three viruses. Currently OPV is routinely given at birth and 6 weeks of age to all infants in South Africa. It replicates in the gut and induces type specific immunity to each of the 3 polioviruses by the eliciting immune response described above. The vaccine strains otherwise known as Sabin OPV 1,2, and 3 differ from the parental wild type strains by critical attenuating mutations in the internal ribosomal entry site (IRES) and capsid (VP1) gene and replicate poorly in the human gut. (There are 57 attenuating mutations in Sabin 1; 2 in Sabin 2 and 10 in Sabin 3.)
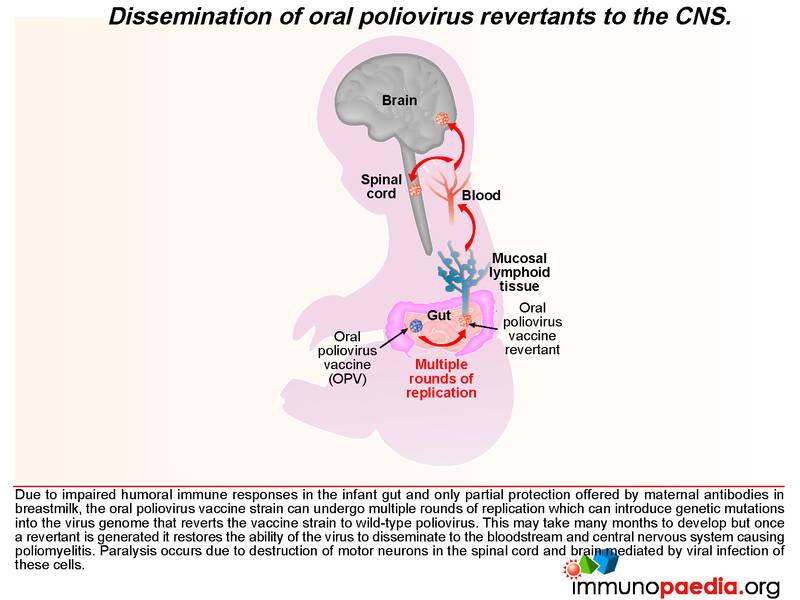
After administering OPV, the vaccine induces cell-mediated immune responses, including antiviral T cell responses and humoral responses that generate mucosal neutralising antibodies. The vaccine strain does not normally disseminate to the central nervous system. However, like all RNA viruses, polioviruses have a high rate of mutation due to lack of proof reading function of the RNA polymerase. Very rarely due to underlying immune dysfunction the vaccine strain can regain virulence due to genetic mutations introduced during continual rounds of replication in the host. This can restore the ability of the virus to disseminate to the central nervous system and cause vaccine-associated poliomyelitis. This gives some clue to what happened in our infant.
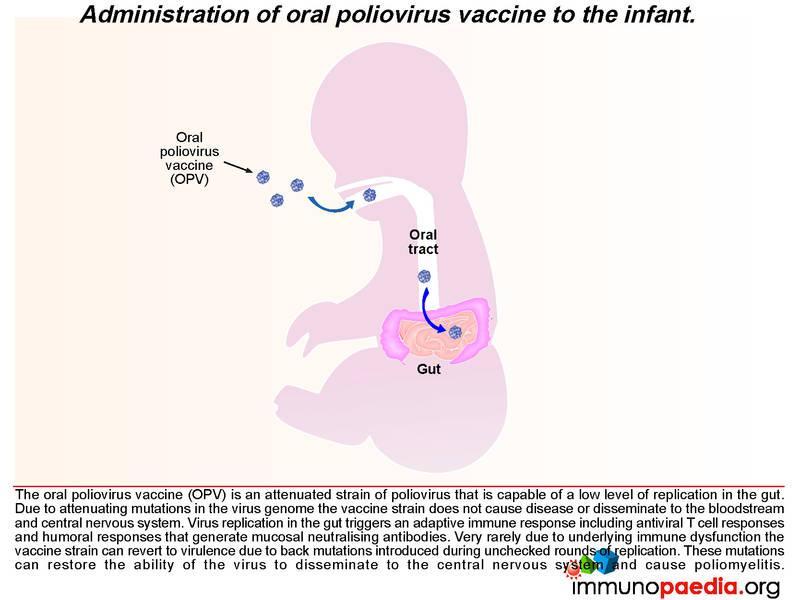 We know in our child that he was agammaglobulinaemic and most likely was unable to mount a humoral response to the replication of the oral poliovirus vaccine. We have described how mucosal antibodies play an important role in the control of enterovirus infections. In such a scenario, the virus is permitted to undergo many rounds of replication, which ultimately can lead to reversion of the vaccine strain to a wild-type phenotype. According to the hospital records, the infant received 2 doses of OPV at birth and at six weeks of age. The virus in his stool had 12nucleotide substitutions relative to Sabin OPV 3 strain and this degree of variation is compatible with a year of replication in his gut. The inability of the child to mount an antibody response allows uncontrolled replication of the vaccine strain of poliovirus.
We know in our child that he was agammaglobulinaemic and most likely was unable to mount a humoral response to the replication of the oral poliovirus vaccine. We have described how mucosal antibodies play an important role in the control of enterovirus infections. In such a scenario, the virus is permitted to undergo many rounds of replication, which ultimately can lead to reversion of the vaccine strain to a wild-type phenotype. According to the hospital records, the infant received 2 doses of OPV at birth and at six weeks of age. The virus in his stool had 12nucleotide substitutions relative to Sabin OPV 3 strain and this degree of variation is compatible with a year of replication in his gut. The inability of the child to mount an antibody response allows uncontrolled replication of the vaccine strain of poliovirus.
 Vaccine associated paralytic poliomyelitis (VAPP)
Vaccine associated paralytic poliomyelitis (VAPP)
This is due to spontaneous neurovirulence of one of the viruses in the OPV. It occurs in approximately 1 in 106 doses of OPV and typically develops within weeks of receiving OPV. The vaccine virus is typically not mutated. Because immunity to enteroviruses is antibody-mediated, patients with B cell immunodeficiencies have a 2000-3000 fold increased risk for VAPP compared with immuncompetent individuals and OPV is contra-indicated in these patients.
Vaccine derived paralytic polio (VDPP)
During replication in the GIT, the attenuating mutations in the vaccine strains are under strong negative selection and readily revert back to wild type. These mutations can result in reversion of the vaccine virus to a virulent phenotype, neuro-invasion and symptomatic disease. This rare complication usually only occurs following prolonged replication in the human gut (more than 6 months) by so called vaccine derived polioviruses (VDPVs). These viruses regain their virulent phenotype through back mutation or recombination with other enteroviruses. They are similar to wild polioviruses in terms of transmissibility and neurovirulence. VDPV may emerge following prolonged replication in either a single individual with a B cell immunodeficiency, or following person to person transmission of vaccine virus in communities with low vaccine coverage. Rarely patients with primary B cell immunodeficiencies may become long-term excretors of OPV. The time taken for this child to present with clinical disease and the number of mutations present in the virus from his stool suggests that this process was occurring in this patient.
Did breastfeeding delay the onset of disease in our patient?
The close temporal association of meningitis/flaccid paralysis with cessation of breastfeeding in our child makes it tempting to speculate that antibodies in breast milk were controlling the viral replication in the gut to low levels and thereby preventing dissemination of the virus to the brain. Infants with agammaglobulinaemia receive partial protection from infection with enteric organisms if they are breastfed due to the presence of maternal immunoglobulins directed towards common gut pathogens. However, their value is limited, and an active mucosal immune response is critical for long-term protection to gut pathogens. Patients with agammaglobulinaemia are particularly prone to enterovirus infections.
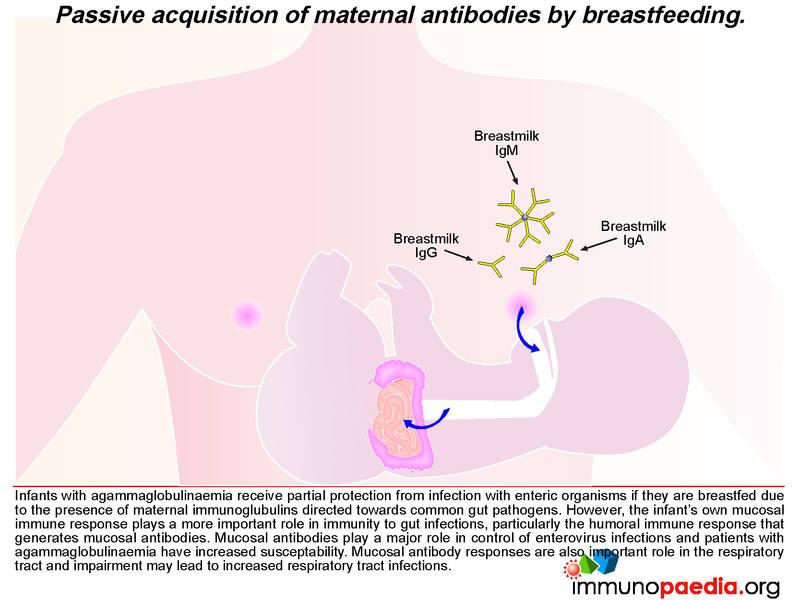 In our case, the presence of antiviral neutralizing antibodies in breast milk may have provided partial protection from dissemination of the poliovirus revertants from the infant’s gut. We thus speculate that shortly after weaning of our infant, the reverted virus was able to replicate more efficiently in the gut, gain access to the bloodstream and disseminate to the central nervous system where infection of motor neurons caused poliomyelitis.
In our case, the presence of antiviral neutralizing antibodies in breast milk may have provided partial protection from dissemination of the poliovirus revertants from the infant’s gut. We thus speculate that shortly after weaning of our infant, the reverted virus was able to replicate more efficiently in the gut, gain access to the bloodstream and disseminate to the central nervous system where infection of motor neurons caused poliomyelitis.
 To know what is causing the agammaglobulinaemia, lets first look at normal B cell development
To know what is causing the agammaglobulinaemia, lets first look at normal B cell development
To recap, in our infant case, we found enough evidence to suggest that the OPV most likely mutated in the gut and reverted to wild type. It is likely that the infant had a primary immunodeficiency, which allowed the reverted poliovirus to cause meningitis and flaccid paralysis. So what may be the reason for the inability to make antibodies?
In the normal process of B cell development in the bone marrow, multipotent haematopoietic stem cells produce lymphoid progenitors, which in turn generate B cell precursors. The development of a B cell with unique antibody specificity requires that the germline DNA coding for the variable part (antigen binding) of the immunoglobulin molecule undergo a programmed set of rearrangements. The process begins at the heavy chain locus of the mu allele: one of a number of D gene segments aligns next to a J gene segment. This is followed by alignment of a V segment to form a composite coding sequence for the variable immunoglobulin domain on the heavy chain. Successful rearrangement allows synthesis of mu heavy chains. The heavy chain pairs with a surrogate light chain (SLC) made of two proteins, delta 14.1 and V preB. Together these form the pre-BCR. Failure to assemble a pre-BCR prompts the B cell to rearrange the other mu heavy chain locus. If this fails, the cell dies.
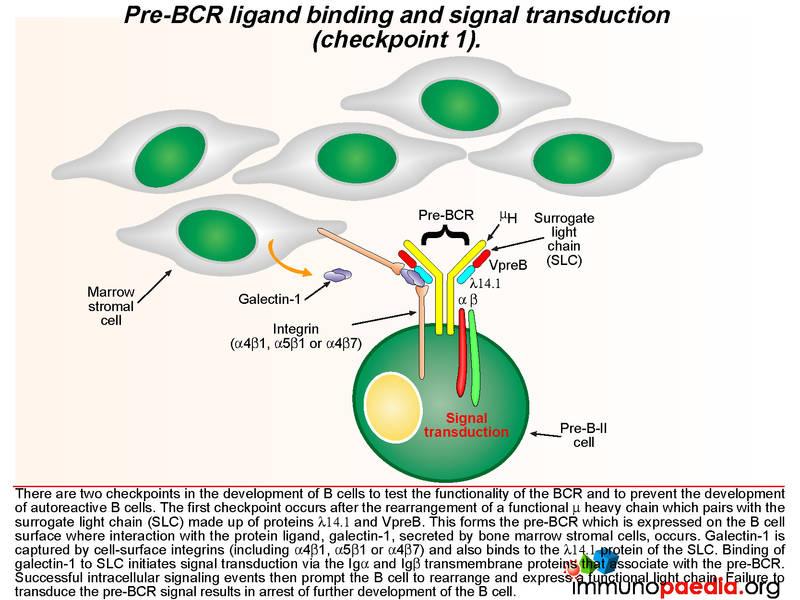
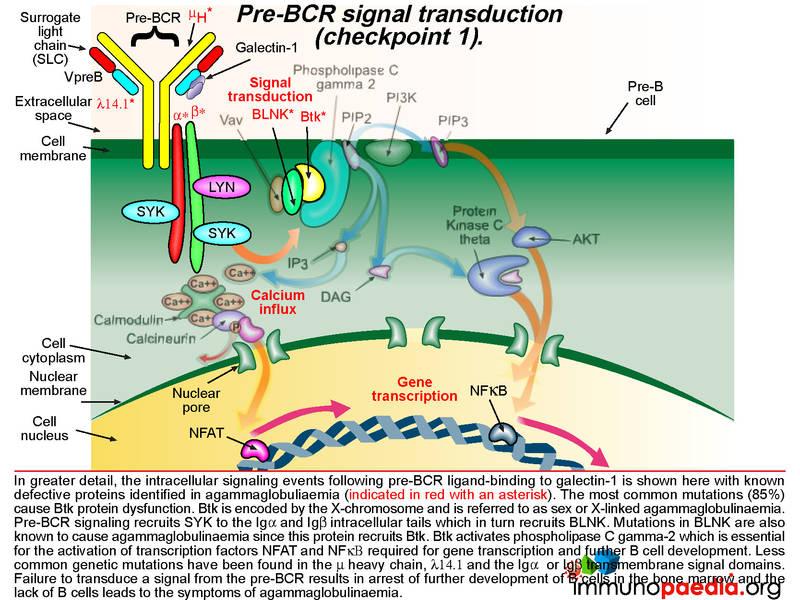
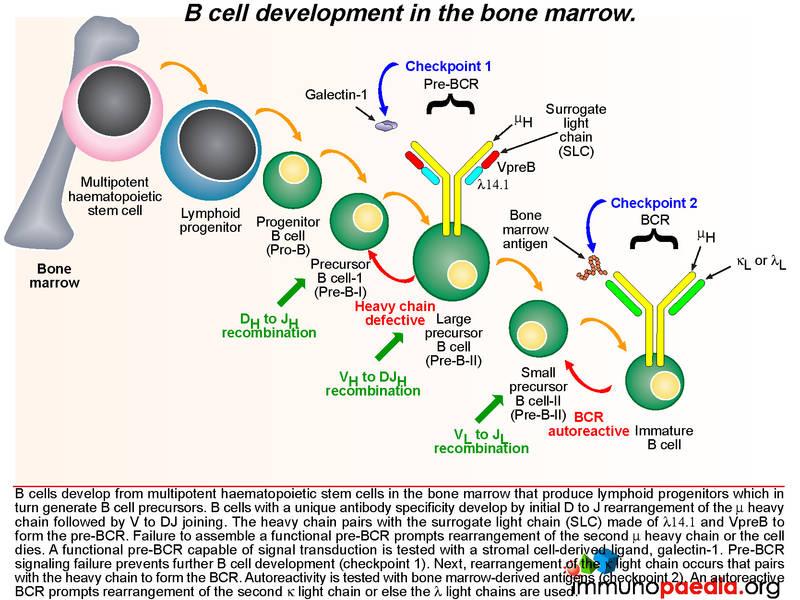 The pre-BCR is then expressed on the B cell surface and is tested for the ability of this receptor to respond to ligand stimulation. This is called checkpoint 1. The ligand is a protein called galectin-1 secreted by bone marrow stromal cells in close contact with the developing B cells. Galectin-1 is captured by integrin molecules on the surface of the B cell and the stromal cells (commonly alpha4beta1, alpha5beta1 or alpha4beta7). Galectin-1 also binds to lambda 14.1 of the SLC and induces intracellular signal transduction via the transmembrane Ig alpha and Ig beta domains that associate with the pre-BCR. Failure of pre-BCR signaling prevents the B cell from further development. This is also the stage of B cell development where defective proteins can lead to agammaglobulinaemia.
The pre-BCR is then expressed on the B cell surface and is tested for the ability of this receptor to respond to ligand stimulation. This is called checkpoint 1. The ligand is a protein called galectin-1 secreted by bone marrow stromal cells in close contact with the developing B cells. Galectin-1 is captured by integrin molecules on the surface of the B cell and the stromal cells (commonly alpha4beta1, alpha5beta1 or alpha4beta7). Galectin-1 also binds to lambda 14.1 of the SLC and induces intracellular signal transduction via the transmembrane Ig alpha and Ig beta domains that associate with the pre-BCR. Failure of pre-BCR signaling prevents the B cell from further development. This is also the stage of B cell development where defective proteins can lead to agammaglobulinaemia.
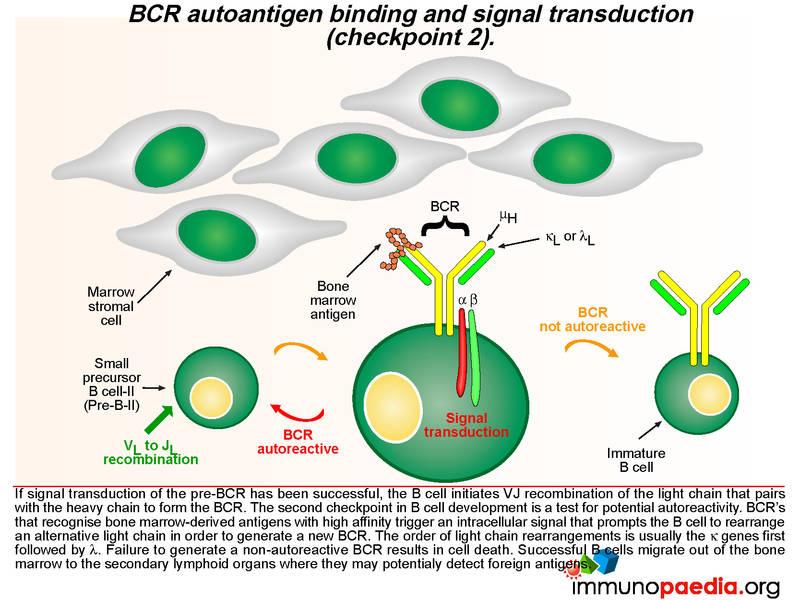
 A successful pre-BCR signal transduction event prompts the B cell to re-arrange the variable region of a kappa light chain gene, which involves V to J joining (there are no D gene segments in the light chain). The light chain replaces the SLC and pairs with the heavy chain to generate a BCR that is expressed on the B cell surface. A second checkpoint occurs at this stage of B cell development to determine whether the newly generated BCR has the potential to bind self-antigens. If the BCR binds with high affinity to bone marrow-derived antigens, an intracellular signal is transduced which prompts the B cell to rearrange the second kappa light chain allele. The new BCR is also tested for autoreactivity. The lamda light chains are used next if the kappa light chains are not successful or else the B cell dies. Successful B cells then migrate out of the bone marrow to the secondary lymphoid organs where they may detect foreign antigens.
A successful pre-BCR signal transduction event prompts the B cell to re-arrange the variable region of a kappa light chain gene, which involves V to J joining (there are no D gene segments in the light chain). The light chain replaces the SLC and pairs with the heavy chain to generate a BCR that is expressed on the B cell surface. A second checkpoint occurs at this stage of B cell development to determine whether the newly generated BCR has the potential to bind self-antigens. If the BCR binds with high affinity to bone marrow-derived antigens, an intracellular signal is transduced which prompts the B cell to rearrange the second kappa light chain allele. The new BCR is also tested for autoreactivity. The lamda light chains are used next if the kappa light chains are not successful or else the B cell dies. Successful B cells then migrate out of the bone marrow to the secondary lymphoid organs where they may detect foreign antigens.
 Lets look at this in greater detail
Lets look at this in greater detail
The cartoon of intracellular signaling events following pre-BCR ligand-binding to galectin-1 shows known defective proteins (indicated in red with an asterisk). The most common genetic mutation (in 85% of cases) causes Btk protein dysfunction. Btk (Bruton tyrosine kinase) maps to the X-chromosome and this type of agammaglobulinaemia is referred to as sex or X-linked (also Bruton’s agammaglobulinaemia). Other less common defective proteins identified include dysfunctional mu heavy chains or lamda 14.1 proteins that are required to complete the pre-BCR. Pre-BCR signaling recruits SYK to the Ig alpha and Ig beta intracellular tails which then recruits BLNK and BLNK recruits Btk. Mutations in Ig alpha and Ig beta and BLNK have been identified Btk is responsible for activating phospholipase C gamma-2, which is essential for the activation of transcription factors NFAT and NFkB required to initiate gene transcription for further B cell development. Failure to transduce a signal from the pre-BCR results in arrest of further development of B cells in the bone marrow and a lack of mature B cells leads to agammaglobulinaemia.
 The second checkpoint in B cell development is also dependent on intracellular cell signaling, but this time the interaction of the BCR with bone marrow-derived antigens is indicative of autoreactivity. Here the B cell is prompted to rearrange an alternative light chain allele, usually in the order of the kappa genes first followed by the lambda genes. In agammaglobulinaemia this stage may not be reached due to failure at checkpoint 1, however, some mutations are “leaky” and permit a reduced level of functionality of the affected proteins. This potentially affects checkpoint 2 where autoreactive B cells may escape elimination due to insufficient signaling of an autoreactive BCR leading to the survival of autoreactive B cells and the potential to develop antibody-mediated autoimmune reactions.
The second checkpoint in B cell development is also dependent on intracellular cell signaling, but this time the interaction of the BCR with bone marrow-derived antigens is indicative of autoreactivity. Here the B cell is prompted to rearrange an alternative light chain allele, usually in the order of the kappa genes first followed by the lambda genes. In agammaglobulinaemia this stage may not be reached due to failure at checkpoint 1, however, some mutations are “leaky” and permit a reduced level of functionality of the affected proteins. This potentially affects checkpoint 2 where autoreactive B cells may escape elimination due to insufficient signaling of an autoreactive BCR leading to the survival of autoreactive B cells and the potential to develop antibody-mediated autoimmune reactions.
Download images for this case
Treatment
High dose IVIG was commenced immediately and the mother was encouraged to recommence lactation.
Clinical Course
The infant improved clinically, but 3 weeks later still had some neurological deficits. He was discharged with an indwelling line for IVIG.
Infection Control
The parents were provided with biohazard bins for disposal of nappies and subsequent incineration, as well as agents to decontaminate the area when nappies were changed in public places.
The stool enterovirus titre dropped over the next few weeks.
Download images for this case
Final Outcome
A confirmed diagnosis of vaccine derived paralytic polio due to X-linked agammaglobulinaemia. The infant was unable to clear vaccine virus because of absent immunoglobulins. This led to ongoing replication of the vaccine virus in the infant’s gut. During this process, the attenuating mutations in the vaccine virus were lost and the virus became neurovirulent.
Download images for this case
References
Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA.(2005).Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annual Reviews Microbiology 59:587-635
Racaniello VR. One hundred years of poliovirus pathogenesis. (2006). Virology. Jan 5;344(1):9-16
Bergelson JM. (2008). New (fluorescent) light on poliovirus entry. Trends Microbiol. Feb;16(2):44-7.
Mårtensson IL, Almqvist N, Grimsholm O, Bernardi AI. (2010) The pre-B cell receptor checkpoint.FEBS Lett. Jun 18;584(12):2572-9
Notarangelo LD., Primary immunodeficiencies. (2010) J Allergy Clin Immunol. Feb;125(2 Suppl 2):S182-94
Mueller S, Wimmer E, Cello J. (2005). Poliovirus and poliomyelitis: a tale of guts, brains, and an accidental event. Virus Res. Aug;111(2):175-93
Download images for this case
Evaluation – Questions & answers
What was the final diagnosis?
A confirmed diagnosis of vaccine associated paralytic polio dues to X-linked agammaglobulinaemia. VAPP occurred due to failure to clear vaccine virus because of absent immunoglobulins. Prolonged passage in the infant’s gut allowed the virus to revert to virulent phenotype and cause neurovirulence.
Explain the difference between VAPP and VDPP?
Vaccine associated paralytic polio occurs within weeks of exposure to OPV. It is due to de novo neurovirulence of one of the 3 viruses in the vaccine (It is not mutated). Individuals with B cell immunodeficiencies have a 2000-3000 fold increased chance of developing this because they can’t control virus replication.
Vaccine derived paralytic polio occurs after prolonged replication in the gut (months to years after exposure to the vaccine). The causative virus contains many mutations. Accumulation of these mutations allows the virus to revert to a virulent phenotype.
Why do you think the infant developed paralysis on cessation of lactation?
Antibodies in breast milk were controlling Polio viral replication in the gut of the infant to low levels and thereby preventing dissemination of the virus to the brain. Infants with agammaglubinaemia receive partial protection from infection with enteric organisms if they are breastfed due to the presence of maternal immunoglubulins directed towards common gut pathogens. The presence of antiviral neutralizing antibodies in breast milk may have provided partial protection from uncontrolled replication of the Poliovirus revertants in the infants’ gut. We thus speculate that after weaning, the reverted virus disseminated to the central nervous system and caused poliomyelitis.
How does OPV work?
There are 3 serotypes of wild poliovirus (poliovirus 1, 2 and 3) and OPV contains attenuated strains of these three viruses. Currently OPV is routinely given at birth and 6 weeks of age to all infants in South Africa. It replicates in the gut and induces type specific immunity to each of the 3 polioviruses by the eliciting immune response described above. The vaccine strains otherwise known as Sabin OPV 1,2, and 3 differ from the parental wild type strains by critical attenuating mutations in the internal ribosomal entry site (IRES) and capsid (VP1) gene and replicate poorly in the human gut. After administering OPV, the vaccine induces cell-mediated immune responses, including antiviral T cell responses, and humoral responses that generate mucosal neutralising antibodies.
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case



