





- Patient presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Evaluation - Questions & answers
- MCQ
- References
Patient presentation
A 5 and half year old female, having recently moved provinces, is seen by a new paediatrician and diagnosed with pneumonia. It is also noted that she is failing to grow consistently and has experienced frequent arrests in weight gain.
Acknowledgement
This case study was kindly provided by Dr Monika Esser MMed Paed, Head of Division of Immunology, N.H.L.S Coastal Branch, Tygerberg Hospital.
History
- Conceived through in vitro fertilisation
- Full term gestation, normal birth weight
- Breastfed until age 2 yrs and 8 month
- She received all recommended infant vaccinations, according to the South African expanded program of immunization (EPI) schedule
- She is the only joint child of her parents, she has 3 adult half-siblings
- All family members are healthy
- According to her mother she has had little contact with other sick children
History Relevant To Infections
- Recurrent otitis media, sinusitis and 3 documented episodes of pneumonia starting from 2 years of age
- At age 2 yrs, due to recurrent infections a low Serum Ig was documented – but no intervention was undertaken
Differential Diagnosis
Primary Immunodeficiency
B cell disorder
- X-linked Agammaglobulinaemia (Bruton Disease)
- IgA Deficiency
- Transient hypogammaglobulinaemia of infancy
- Hypogammaglobulinaemia (Common Variable Immunodeficiency)
- Ataxia Telangiectasia
- Ataxia Telangiectasia like disorder (ATLD)
- Nijmegen breakage syndrome (NBS)
T cell disorder
- Severe combined immunodeficiency
- Hyper-IgM syndrome
Cystic Fibrosis – common genetic disorder in SA white population, leading to respiratory infections and failure to thrive
- Cerebral Palsy
- Atopy – common undiagnosed cause of recurrent URT symptoms
- Poverty – early exposure to overcrowding or viruses resulting in perinatal respiratory problems
- HIV
Examination
- Age 5 years, 5 months – slender build – weight: 16.6kg, height 105 cm, both on 10th centile for age.
- Moderate generalised lymphadenopathy.
- Nasal speech and slurring of words noted – mother claims onset related to adenoidectomy and tonsillectomy at age 3 and has been receiving speech therapy.
- Crepitations heard in left lung base – was hospitalised 5 days previously for pneumonia.
- Grommets in situ (3rd set).
Investigations
| Value | Normal Limits | |
|---|---|---|
| FBC | ||
| WBC | 22.4 x 109/l | |
| Relative Lymphopaenia | ||
| Hb | 12.8/l | |
| MCV | 81 | |
| Platelets | Normal | |
| Sweat test | Normal | |
| Serum Ig’s | ||
| IgG | 0.33g/l | 6 - 16g/l |
| IgA | 0.07g/l | 0.6 - 3g/l |
| IgM | 1.63g/l | 0.5 - 2.6g/l |
| Results prior to first intravenous immunoglobulin (IVIg) | ||
| CD3 | 1371 (83%) | 1800 - 3000 |
| CD4 | 558 (33%) | 1000 – 1800 |
| Ratio | 0.67 | |
| CD8 | 817 (49%) | 800 – 1500 |
| CD19 | 133 (8%) | 700 – 1300 |
| NK | 163 (10%) | 200 – 600 |
| Immunoglobulin Studies | ||
| IgA | 0.49 | (0.70 – 4.0g/l |
| IgM | 2.3199999999999998 | 0.40 – 2.3g/l |
| IgG | 7.54 | 7.0 – 16.0g/l |
| Specific Antibodies | ||
| Tetanus antibodies (protein ag) | 3.27IU/ml (N) | |
| Antibodies to Streptococcus pneumoniae IgG (polysaccharide Ag) | 48.19mg/L (low N) |
Discussion
Management and treatment
Bruton like Agammaglobulinaemia was excluded because CD19 (B Cells), IgM and IgA are all present.
Due to the preservation of IgM – CD40 ligand assay was requested which was normal as demonstrated with normal binding of CD154. CD40 is expressed on B cells, macrophages, dendritic cells, and endothelial cells essential for activation. In B cells the interaction with CD40 cell ligand is needed for CLASS SWITCHING for Ig production other than IgM or IgD, ie co – stimulatory binding leads to T cell dependent antibody production (IgG, IgA or IgE).
A diagnosis of Hypogammaglobulinemia was made with low but adequate B cells (greater than 2%) and serum IgG (Se IgG) and IgA below standard deviations for age. Differentiating between primary (congenital) and secondary Hypogammaglobulinemia is important as this has implications for evaluation and management of the patient. Secondary Hypogammaglobulinemia was ruled out as there was no indication of immunosuppressive drugs in the patient history and no indication of disease that leads to decreased antibody production or increased antibody loss(Otani et al., 2022). Transient Hypogammaglobulinemia was also considered but ruled out because she is more than 2 years old.
Pathophysiology
What is the main vulnerability of patients with low antibody levels? Antibodies are required to clear extracellular bacteria. These bacteria have capsules that are not recognised by phagocytic cells of the immune system. To clear such bacteria, humans rely on antibodies, together with complement, to assist phagocytic cells to recognise and ingest the bacteria. There are also some viruses that require antibodies for control. They have low lymphocyte phenotypes, except CD8. Although the levels are low there are antibody responses present to proteins and polysaccharides.
 They have low lymphocyte phenotypes, except CD8 . Although the levels are low there are antibody responses present to proteins and polysaccharides.
They have low lymphocyte phenotypes, except CD8 . Although the levels are low there are antibody responses present to proteins and polysaccharides.
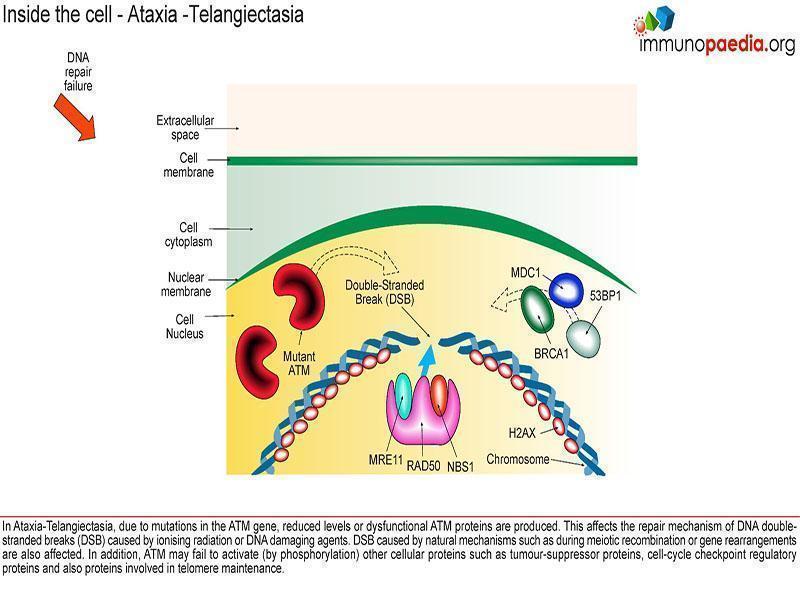
Other Causes of Hypogammaglobulinemia
Secondary to:
- Decreased production
- Malignancy (lymphoma, thymoma, leukaemia, multiple myeloma)
- Medications (carbazepine, oxcarbazepine, immunosuppressive agents)
- Infection (e.g. paediatric HIV)
- Starvation
- Increased catabolism or loss
- Protein-losing enteropathy
- Chylothorax
- Hypercatabolic states
Clinical Course
- Regular intravenous immunoglobulin (IVIG) instituted.
- Follow up with the same paediatrician who monitored the course and Serum IgG (SeIgG) levels.
Follow up visit at 6 years and 7 months
- Patient is receiving 12 grams IVIG every 3 weeks with trough levels of IgG, 7-8 grams.
- No further significant infections reported.
- CT Scan shows lungs clear.
- Weight 19kg and height 110cm.
- Chest is clear on auscultation.
- Clean perforations in both ears with hearing loss noted on the left side.
Next follow up visit at 7 years and 5 months
- Patient has remained infection free, but balance problems mentioned by mother.
- She has had difficulty with head control and is receiving Occupational therapy.
- Urgent referral to Neurology – Ataxia diagnosed
- Alpha Fetoprotein elevated
- New diagnosis made
Download images for this case
Evaluation – Questions & answers
What is the diagnosis?
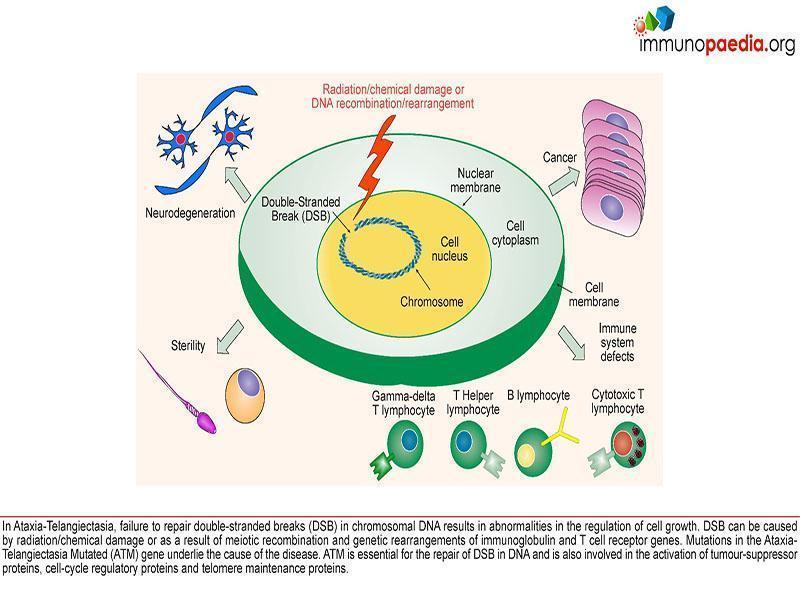
Ataxia and Immunodeficiency without Telangiectasia (AT). Ataxia-telangiectasia (AT) is an autosomal recessive genetic disorder. The characteristic features are immune dysfunction, progressive neurodegeneration (cerebellar degeneration),
cutaneous abnormalities (incl. telangiectasia ), radiosensitivity, sterility and cancer predisposition. Children with AT have deficiencies in both cellular and
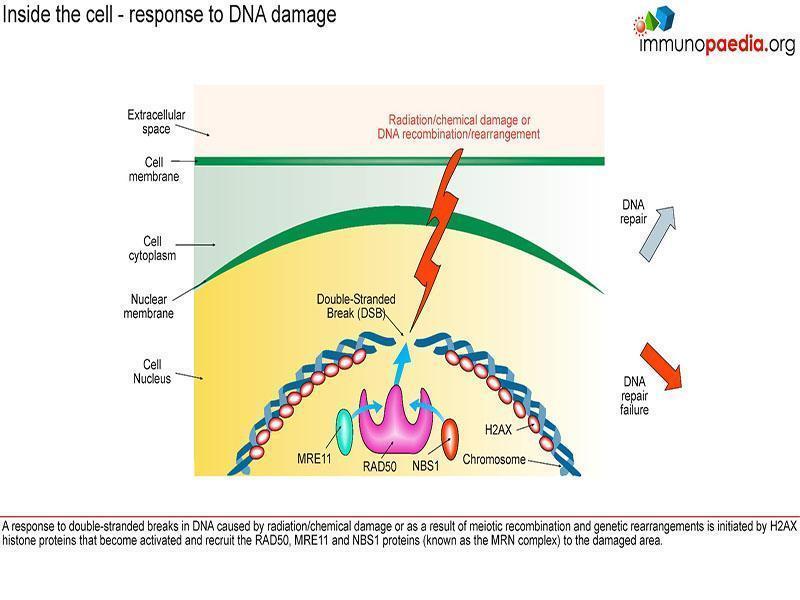
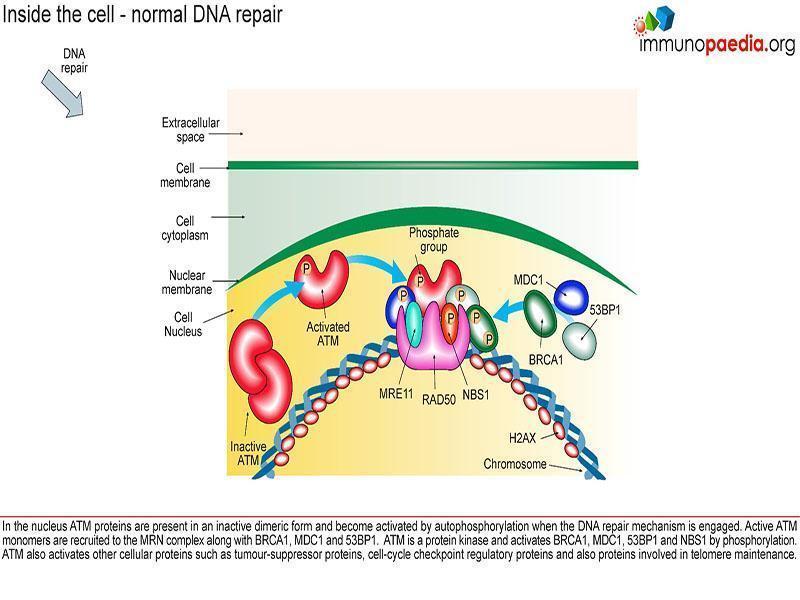
humoral immunity including reduced levels of IgA, IgE and IgG2, absolute lymphopenia and a decreased ratio of CD4+ helper cells to CD8+ suppressor T cells. There is increased susceptibility to recurrent upper and lower respiratory tract infections (particularly those caused by encapsulated bacteria) and an increased risk for the development of malignancies. ATM, the gene associated with AT has been identified as a single mutation on the long arm of chromosome 11 at 11q22-23. This gene controls the production of phosphatidylinositol 3/4 kinase family, an enzyme which is a crucial nexus for the cellular response to DNA double-stranded breaks. So, these kinases are important players in the cellular responses that prevent cancer, control DNA damage and control cellular
responses to stress. The ATM gene is also involved in double strand break repair mechanisms in physiological conditions such as meiotic recombination, the assembly of the T cell receptor and immunoglobulin (Ig) genes by V(D)J recombination and efficient class switch recombination(Amirifar et al., 2019). Accordingly, AT is thus a disease that results from defects in the response to specific types of DNA damage, which is related to pathogenesis including immune dysfunction, lymphoma predisposition and sterility(Amirifar et al., 2019).
Classification
There appears to be three main forms of AT:
- Pure AT where patients present with all/most of the diagnostic symptoms.
- Attenuated AT where sufferers do not possess all of the diagnostic symptoms.
- Carrier AT where individuals with a single ATM mutation show an increased risk of cancer.
These are sometimes classified into ‘types’ from I to IV:
- Type I: is the classic syndrome with all manifestations.
- Type II: lacks some of the typical findings but shows radiosensitivity.
- Type III: has the classic clinical findings but is not radiosensitive.
- Type IV: shows only some clinical features and is not radiosensitive.
Carrier detection and prenatal diagnosis are possible at specialized centres. In rare instances patients with milder manifestations of the clinical or cellular characteristics of the disease have been reported and have been designated “AT variants.” Variant forms may have a detectable level of functional ATM while exhibiting some of the cellular features of AT.
Pathology and Outcome
Primary Immunodeficiency is variable with variable humoral deficiency and thymic hypoplasia. Increased susceptibility to recurrent upper and lower
respiratory tract infections, particularly those caused by encapsulated bacteria. Historically bacterial pneumonia and chronic pulmonary disease have been the major cause of death with the second most common cause being cancer (10-30% lifetime prevalence)(Amirifar et al., 2019). Diffuse Cortical Degeneration of cerebellum – reflecting progressive loss of Purkinje and granular cells is most striking. Furthermore, patients with class switching recombinant disorder (CSRD) have a more serve course of disease leading to a lower quality of life and shorter survival in early ages than other AT patients(Amirifar et al., 2019).
How is Ataxia telangiectasia (AT) diagnosed?
AT is diagnosed clinically and with laboratory tests:
- Early-onset progressive cerebellar ataxia (earliest sign)
- Occulo-cutaneous telangiectasia (dilated blood vessels in the eyes and skin) at age 3-6 yrs.
- Immunodeficiency mostly through lowering of IgA, IgG and IgE levels. In AT the humoral immunodeficiency may not be significant enough to require IVIG and the cellular deficiency rarely gives rise to significant infections other than chronic and recurrent warts.
- Chromosomal instability
- Hypersensitivity to ionising radiation
- Increased incidence of malignancies primarily lymphoid (Non-Hodgkin’s lymphoma).
- Raised alpha-fetoprotein levels (differential after 8/12 of age – Hepatitis, Hereditary Tyrosinaemia, Hepatoblastoma or asymptomatic hereditary persistence).
- Gene too large for practical screening.
What is the main effect of AT on the immune system?
Why are IgM isotype levels not affected in AT?
What is the main vulnerability of patients with low antibody levels?
What is CD40 ligand?
What is immunoglobulin class switching?
What is IVIg therapy?
What is hypogammaglobulinaemia?
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case
- Amirifar, P., Ranjouri, M. R., Yazdani, R., Abolhassani, H., & Aghamohammadi, A. (2019). Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatric Allergy and Immunology, 30(3), 277–288. https://doi.org/10.1111/PAI.13020
- Otani, I. M., Lehman, H. K., Jongco, A. M., Tsao, L. R., Azar, A. E., Tarrant, T. K., Engel, E., Walter, J. E., Truong, T. Q., Khan, D. A., Ballow, M., Cunningham-Rundles, C., Lu, H., Kwan, M., & Barmettler, S. (2022). Practical guidance for the diagnosis and management of secondary hypogammaglobulinemia: A Work Group Report of the AAAAI Primary Immunodeficiency and Altered Immune Response Committees. Journal of Allergy and Clinical Immunology, 149(5), 1525–1560. https://doi.org/10.1016/J.JACI.2022.01.025
- Viallard, J. F. (2023). Conduite à tenir devant une hypogammaglobulinémie. La Revue de Médecine Interne, 44(3), 133–138. https://doi.org/10.1016/J.REVMED.2023.01.010
Download images for this case



