





Introduction
- The immune system’s three basic functions are Defence, Surveillance and Homeostasis
- Elaborate and redundant tolerance mechanisms are in effect both during maturation and later in the lymphocyte life-cycle, leading to clonal deletion or anergy
- Anergy can be reversed to allow recruitment of autoreactive clones to maximise receptor diversity (e.g. during infection)
- Autoimmunity (Table 1) emerges when self-tolerance mechanisms fail – infections can lead to breaking of tolerance [Case Study – A 9 year old girl presents with body swelling, shortness of breath and backache, Case Study – Why can I not walk today?]
- Primary immunodeficiencies can also present with altered surveillance (leading to malignancy) or altered homeostasis (leading to autoimmunity)
Table 1. Partial list of autoimmune diseases
| Disease | Main organ affected | Proposed self-antigen(s) | Clinical presentation |
|---|---|---|---|
| Organ-specific autoimmune diseases | |||
| Multiple sclerosis | Central nervous system | Myelin basic protein, myelin oligodendrocyte protein | Loss of vision, weakness of limbs, sensory abnormalities, incontinence |
| Sympathetic ophthalmia | Eye | Various uveal antigens | Eye pain, loss of vision, sensitivity to light |
| Graves' disease | Thyroid | Thyrotropin receptor | Hyperthyroidism (weight loss, nervousness, palpitations, diarrhea), exophthalmos |
| Hashimoto's thyroiditis | Thyroid | Thyroperoxidase, thyroglobulin | Hypothyroidism (weight gain, constipation, skin changes, myxedematous dementia) |
| Goodpasture's syndrome | Lung, kidney | Glomerular basement membrane (type IV collagen) | Kidney and respiratory insufficiency |
| Pernicious anemia | Stomach | Intrinsic factor | Anemia, gastritis |
| Crohn's disease * | Intestine | ? microbial antigens | Hemorrhagic diarrhea, abdominal pain, draining fistulas |
| Ulcerative colitis * | Large Intestine | ? microbial antigens | Hemorrhagic diarrhea, abdominal pain |
| Diabetes mellitus type I | Pancreas | Islet cell, insulin, glutamic acid decarboxylase (GAD) | Polyphagia, polyuria, polydipsia, weight loss |
| Immune thrombocytopenia | Platelets | Glycoproteins on the surface of platelets | Easy bruising, hemorrhage |
| Myasthenia gravis | Muscle | Acetylcholine receptor | Muscle weakness, fatiguability |
| Hemolytic anemia | Red cells | I antigen | Anemia |
| Systemic autoimmune diseases | |||
| Sjögren's syndrome | Salivary and lacrimal glands | Nuclear antigens (SSA, SSB) | Dry eyes, dry mouth, lung and kidney disease |
| Rheumatoid arthritis | Joints, lung, nerves | Citrulinated peptides in the joint, IgG | Deforming arthritis, skin nodules, occasional lung and nerve involvement |
| Wegener's granulomatosis | Lung, kidney | Proteinase 3 (c-ANCA) | Sinusitis, shortness of breath, kidney failure |
| Systemic lupus erythematosus | Kidney, skin, joints, central nervous system | DNA, histones, ribonucleoproteins | Arthritis, skin rashes, kidney insufficiency, nerve damage |
- Autoimmunity affects ~5% of the population, predominantly women in their reproductive years, with peak incidences during adolescence and the fourth through fifth decades
- An understanding of lymphocyte physiology and tolerance mechanisms largely derives from transgenic animal models, as well as genomics and proteomics research
- There has been some success translating such observations to humans, but additional clinical studies are required, including to better understand the pathogenesis of autoimmunity
- Improved knowledge may be applied to generate more efficacious and safer autoimmune therapies
Organ-specific vs. Systemic Autoimmunity
- Organ-specific autoimmunity is targeted against a single organ while systemic autoimmunity affects diverse tissues
- It is not uncommon for a patient to have symptoms of more than one autoimmune disease (known as ‘overlap’, or undifferentiated collagen vascular syndrome)
- In addition, a patient with one autoimmune disease may have serologic markers – but no clinical manifestations – of another
- An autoimmune response is defined as demonstration of autoantibodies or T-cell autoreactivity, which may or may not be associated with clinically manifested autoimmune disease
- Many healthy women have serum antinuclear antibodies, but demonstrate no symptoms of SLE (whose diagnosis requires the presence of clinical features such as rash, arthritis, and renal involvement)
- Although underlying molecular etiologies remain elusive for most autoimmune diseases, it is thought that autoimmunity is multifactorial, resulting from a complex interplay between genetic susceptibility, environmental triggers, and aberrant immune regulation [Table 2, Figure 2]
Table 2. Aetiology of autoimmune diseases
| Genetic factors |
|---|
| MHC |
| Non-MHC genes |
| Environment |
| Microorganisms |
| Viruses |
| Trauma |
| UV light |
| Immune response dysregulation |
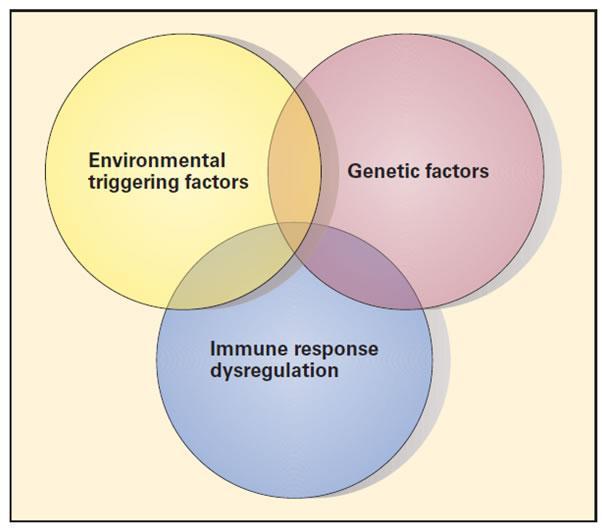
Figure 2. Schematic representation of the three etiopathogenic factors involved in autoimmune diseases. The figure illustrates the complex interplay between genetic factors, environmental triggers, and regulatory aberrations of the immune response responsible for the development of autoimmune disease [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Aetiology: Genetics
- There is ample evidence for genetic susceptibility (e.g. clustering in families, higher concordance in monozygotic twins, higher prevalence in specific ethnic groups, etc.)
- Rarely, single gene mutations lead to autoimmunity (monogenic diseases, e.g. ALPS), but most autoimmune diseases are polygenic (30+ genes contribute)
- In animal models, a single gene mutation can lead to different outcomes, depending on the rest of the genetic make-up
- The first genes shown to have a strong association with many autoimmune diseases are within the MHC locus [Table 3]
Table 3. Association of autoimmune diseases and MHC
| MHC-I association |
|---|
| Ankylosing spondylitis (HLA-B27) |
| Reactive arthritis (HLA-B27) |
| Psoriasis (HLA B-13, B-16, B-17) |
| MHC-II association |
| Systemic diseases |
| Systemic lupus erythematosus (HLA-DR2 and DR3) |
| Rheumatoid arthritis (HLA-DR4) |
| Organ-specific diseases |
| Type 1 diabetes mellitus (HLA-DR3 and DR4) |
| Multiple sclerosis (HLA-DR2) |
- MHC-I-associated diseases include a cluster of newly-classified diseases: the autoinflammatory diseases (including the spondyloarthropathies, such as ankylosing spondylitis [Case Study – 14 year old with severe hip pain], reactive arthritis, and psoriasis)
- 90% of ankylosing spondylitis patients have the HLA-B27 gene, but not all people with this gene develop the disorder (precise predisposing mechanisms are not yet known, but animals transgenic for this gene develop spontaneous autoimmunity with a similar disease pattern)
- Many more autoimmune diseases are associated with MHC-II than with MHC-I, including SLE (HLA-DR2, DR3), RA (HLA-DR4), type 1 DM (HLA-DR3, DR4), and MS (HLA-DR2) [Table 3]
- HLA-DRB1*0401 and 0404 are associated with more severe forms of rheumatoid arthritis
- Certain HLA alleles can also predispose to medication hypersensitivities [Case Study – A case of acute fever, rash and vomiting (Abacavir hypersensitivity)]
- Other immune genes (e.g. those encoding components of the complement system or TNF family cytokines) are also located in the same chromosomal region and may further account for its association with autoimmunity
- Genetic associations outside the MHC locus include the autoinflammatory disorders (e.g. genes for FcγR have a moderate association with SLE)
- Improved understanding has led to reclassification of many diseases originally associated with adaptive immunity as disorders of innate immunity (e.g. autoinflammatory diseases)
- Cronh’s disease was previously linked to overactive Th1 and Th17 mucosal responses, but is now known to have genetic abnormalities (e.g. sequence variants of NOD2/CARD15) predisposing to defective innate signalling pathways and thus inadequate immunity
- NOD2/CARD15 and NFκB activation may be key to induction of intestinal mucosal inflammation and may be necessary for maintenance of gut homeostasis
- Mucosal epithelia and macrophages could be the locus of the primary pathological event, with T-cell activation as a secondary effect leading to chronic inflammation, perhaps to compensate for inadequate innate responses
Aetiology: Environmental
- Geographic differences in prevalence and severity of autoimmunity (e.g. lower rates of autoimmunity at lower latitudes) support a role for environmental triggers
- In most cases, the causal relationship between environmental factors and autoimmune diseases has not yet been firmly established
- However, the most characteristic example is rheumatic fever: infection with certain streptococci leads to formation of antibodies cross-reactive with bacterial and host joint/heart antigens, and thus autoimmune endocarditis
- This phenomenon is known as ‘molecular mimicry’ [Case Study – A case of swollen hands]- acute post-streptococcal glomerulonephritis) although this is probably rather mediated by immune complex deposition
- Damage to tissues by microbes, trauma, and UV radiation can also lead to exposure of previously-sequestred autoantigens, as discussed with regards to sympathetic ophthalmia
- Chronic myocarditis and hepatitis post-infection with Coxsackie or hepatitis C virus are thought to be partly due to autoimmunity against previously sequestered heart or liver antigens
- Exposure to sunlight can also exacerbate SLE, as damaged keratinocytes release nuclear autoantigens
- Drugs (e.g. hydralazine, procainamide, isoniazid) can also induce an SLE-like syndrome called drug-induced lupus erythematosus, possibly via altering the methylation status of pro-inflammatory genes
Breaking Tolerance
- Since this usually occurs as a consecutive series of many events (rarely due to a single genetic/environmental factor), breaking of tolerance can be conceptualised as being set in motion by just the right stimuli occurring against the backdrop of a predisposing immunological milieu
- This milieu is infleunced by genetics, prior antigen encounter, local factors in target organs, and other endogenous factors (e.g. the immune-modulating effects of hormones such as cortisol and oestrogen)
- Autoreactive T-cells can, for example, proliferate peripherally during an infection or on release of previously-sequestered antigens, or defects of apoptosis can facilitate development of T-cell autoreactivity
- Similar mechanisms can lead to proliferation of autoreactive B-cells
- Such events collectively lead to dysregulation and loss of tolerance
Diseases of Dysregulation
- There are four broad groups of immune regulation disorders (Table 4), which present with increased susceptibility to infection, lymphoproliferative disorders, and autoimmunity
- McKusick’s catalog of Mendelian Inheritance in Man (MIM) assigns numerical codes to a comprehensive list of inherited diseases, genes and functional DNA segments
- In the MIM system, an initial digit of 1 indicates an autosomal dominant Mendelian trait; 2 indicates autosomal recessive; and 3 indicates X-linked
- The first group of disorders (immunodeficiency with hypopigmentation), includes conditions characterised by phagocytic cell defects
- An example is Chediak-Higashi Syndrome (CHS), due to mutation of the lysosomal trafficking regulator gene, leading to impaired melanin transport and giant neutrophil granules
- The resulting immunodeficiency leads to increased risk of intracellular bacterial infection and albinism
Table 4. Summary of the defects of immune dysregulation comparing multiple features of genetics and inheritancepatterns and molecular defects with clinical manifestations of disease susceptibility
| Group | Direction of IM effect | Mechanism of action | Example of IM agents | Healthy or diseased subjects who receive immunomodulators |
|---|---|---|---|---|
| Group 1 | ||||
| Immunopotentiators | ↑ | No requirement for antigenic specificity | Adjuvants, hormones (e.g., vitamin D) | Healthy subjects receiving vaccines for prevention of infectious diseases |
| Subgroup 1a nonspecific | ||||
| Subgroup 1b specific | Require antigenic specificity | Vaccines, polyclonal antibodies, monoclonal antibodies | Antibody replacement therapy for patients with humoral immunodeficiency | |
| Group 2 | ||||
| Immunosuppressants | ↓ | Produce broad-based immunosuppression at multiple sites | Irradiation, cytotoxic drugs, glucocorticoids, immunophilins (e.g., cyclosporine), intravenous immunoglobulin (IVIG) | Subjects with autoimmune disorders, allergic diseases, and cancer |
| Subgroup 2a nonspecific | ||||
| Subgroup 2b specific | Target specific cells, cytokines, or receptors | Therapeutic polyclonal and monoclonal antibodies (e.g., anti-TNF agents), therapeutic fusion proteins (e.g., etanercept), soluble receptor constructs (e.g., anakinra), cytokines (e.g., interferons) | ||
| Group 3 | ||||
| Tolerance-inducing agents or procedures | ↓ | Directed at provoking immune tolerance by involvement of Treg cells, immunosuppressive cytokines (e.g., TGF-β, IL-10), or other mechanisms | Allergy immunotherapy (SCIT and SLIT) | Subjects with allergic disease |
The Second Group of Disorders
- The second group of disorders (familial haemophagocytic lymphohistiocytosis [HLH] syndromes), includes conditions characterised by defects of cell-mediated cytotoxicity and thus susceptibility to viral infection and malignancy [Case Study – A case of decreased joint function, fever and rash]
- The collection of disorders is characterised by proliferation and infiltration of hyperactive macrophages and T-cells, leading to destruction of erythrocytes by macrophages which have been triggered by excessive lymphocyte-derived cytokine production
- HLH comprises inherited and acquired defects, including mutations in genes encoding molecules involved in cytotoxicity (e.g. of macrophages, CTL, NK cells, and NK T-cells), and defects resulting from malignancy, infections (e.g. EBV, HIV), and autoimmunity
- Five primary disease subtypes (the FHL1-5 disorders) are differentiated based on genetic loci and where in the cytotoxic pathway the defect occurs
- Cytotoxicity depends on assembly of perforin into a transmembrane pore on the target cell, into which granzymes are secreted to induce apoptotic cell death (thus, defects can occur at various levels , including cell-cell contact, perforin, granzyme transport, etc.)
- FHL2: defects in perforin pore formation
- FHL3: defective transport and trafficking of granzyme
- FHL4: defective fusion of cytotoxic cells and their targets
- Inability to extinguish inflammatory reactions leads to prolonged, excessive production of Th1-type pro-inflammatory cytokines (especially IFNγ)
- HLH syndromes can result in a life-threatening ‘accelerated phase’, which manifests as susceptibility to viral infection, enhanced macrophage activity, or haemophagocytosis, leading clinically to prolonged fever, hepatosplenomegaly, neurologic abnormalities, and cytopenia [Case Study – A case of persistant hectic fever]
The Third Group of Disorders
- The third group of disorders (lymphoproliferative syndromes) involves defects of signalling pathways normally responsible for viral clearance
- This leads to severe mononucleosis, HLH secondary to viral infection, hypogammaglobulinaemia, and lymphoma
- Lymphoproliferative syndromes comprise three distinct clinical presentations, each triggered by EBV infection:
- 1) X-linked lymphoproliferative disorder 1 (XLP1), where the affected gene, SH2D1A, encodes adaptor proteins involved in intracellular signalling in cytotoxic lymphocytes, profoundly increasing EBV susceptibility
- This rare immunodeficiency (with lymphohistiocytosis, hypogammaglobulinaemia, and lymphomas) usually develops in males in response to EBV infection and can lead to fulminant mononucleosis syndrome (rapid deterioration with hepatitis, anaemia, thrombocytopenia) or virus-associated haemophagocytoic syndrome, which kills ~70% of XLP patients before the age of 10
- Those who do not develop acute fulminant disease will develop hypogammaglobulinaemia, aplastic anaemia, and lymphoma
- Other herpes viruses may also trigger acute mononucleosis syndrome
- 2) X-linked lymphoproliferative disorder 1 (XLP2), where the X-linked inhibitor of apoptosis (XIAP) gene is mutated
- This enhances lymphocyte apoptosis in response to various stimuli (e.g. ligation of the TCR-CD3 complex, Fas, and TRAIL receptor), and leads to lower numbers of NK T-cells (XIAP is required for survival and differentiation of NK T-cells)
- Longer-term consequences (besides acute mononucleosis) can occur even without EBV infection, so bone marrow transplant should be considered early
- 3) An autosomal recessive form, with a homozygous mutation in the IL-2-inducible T-cell kinase gene (ITK)
- All three forms share lack of NK T-cells, suggesting that they play a role in resistance to EBV infection [Case Study – An unusual cause of fulminant hepatitis]
- 1) X-linked lymphoproliferative disorder 1 (XLP1), where the affected gene, SH2D1A, encodes adaptor proteins involved in intracellular signalling in cytotoxic lymphocytes, profoundly increasing EBV susceptibility
The Fourth Group of Disorders
- The fourth group of disorders includes several entities associated with autoimmunity resulting from defective apoptosis and impaired Treg activity
- This group includes ALPS, APECED, IPEX, and CD25-deficiency
- ALPS refers to defects in apoptosis (Ia is due to mutations in Fas, Ib is due to mutations in FasL, II is due to caspase 8 and 10 defects, and III refers to phenotypes without known genetic causes)
- Apoptosis normally prevents disastrous acculumation of cells, and thus autoimmunity (e.g. TTP, haemolytic anaemia) and lymphadenopathy
- The Fas-FasL interaction normally leads to intracellular caspase activation and apoptosis, but Fas mutations disrupt normal Fas trimer oligomerisation, leading to deranged lymphocyte apoptosis (Table 5)
- Fas mutations have variable penetrance
- Infections are not part of the ALPS phenotype, unless due to immunosuppressive therapy or as a complication of splenectomy, but the risk of lymphoma is greatly elevated
- Characteristic of this, is the reduced number of double-negative (CD4-CD8-) αβT-cells
Table 5. Clinical and genetic features of the autoimmune polyendocrine syndromes.
| Feature | Autoimmune polyendocrine syndromes (APS) | ||
|---|---|---|---|
| Autoimmune polyendocrine syndrome type I (APSI) or (APECED) | Autoimmune polyendocrine syndrome type II (Schmidt's syndrome) | Immune dysregulation, polyendocrinopathy, enteropathy (X-linked) (IPEX) syndrome | |
| Prevalence | Rare | Common | Very rare |
| Time of onset | Infancy | Infancy through adulthood | Neonatal period |
| Gene and inheritance | AIRE (on chromosome 21, recessive) | Polygenic | FOXP3 , X-linked |
| HLA genotype | Diabetes (risk decreased with HLA-DQ6) | HLA-DQ2 and HLA-DQ8; HLA-DRB1*0404 | No association |
| Immunodeficiency | Asplenism, susceptibility to candidiasis | None | Overwhelming autoimmunity, loss of regulatory T cells |
| Association with diabetes | Yes (in 18%) | Yes (in 20%) | Yes (in majority) |
| Common phenotype | Candidiasis, hypoparathyroidism, Addison's disease | Addison's disease, type 1A, diabetes, chronic thyroiditis | Neonatal diabetes, malabsorption |
Autoimmune Poly-endocrine Syndromes
- This heterogeneous group of diseases (APS1 and 2, and IPEX) displays autoimmunity against more than one endocrine organ (non-endocrine organs can also be affected) [Table 5]
- An array of clinical features is possible, but the commonest are hypoparathyroidism, candidiasis, and adrenal insufficiency
- APS1 (aka APECED): mild immune deficiency with persistent mucosal and cutaneous candidiasis, parathyroid dysfunction leading to hypocalcaemia, and Addision’s disease leading to hypoglycemia and hypotension
- The aetiology is a monogenic defect in AIRE (important in central tolerance to endocrine antigens) [Figure 3]
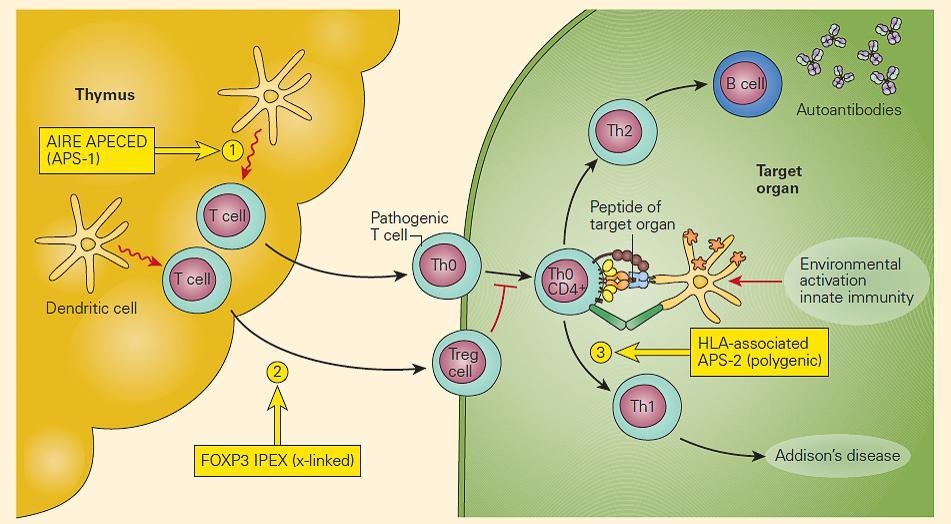
Figure 1. Schematic representation of the molecular defects associated with the autoimmune polyendocrine syndromes. Deficiency of AIRE in the thymic epithelial or DC favors the release of autoreactive pathogenic T cells to the periphery, resulting in APECED (#1); mutations in FOXP3 on T cells impair the development of Treg cells, leading to the IPEX syndrome (#2); APS-2 is a polygenic defect and has been associated with certain HLA phenotypes (#3). (Adapted with permission from Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med. 2004;350:2068–79.). [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
- APS2 (aka Schmidt’s Syndrome): often insidious and more heterogeneous, with features including hypothyroidism, Addison’s, and diabetes (there may also be vitiligo and a history of intermittent severe hypoglycaemia and fatigue)
- Etiologically polygenic: precise mutations are not identified, but risk factors are HLA types DQ2, DQ8, and DRB1*0404
- IPEX (Figure 2): the most rare and serious autoimmune poly-endocrine syndrome, due to Foxp3 mutation leading to and absence of Treg (although a third of cases do not have Foxp3 mutations) and poor regulation of cytokine production
- Every antigen encounter requires both lymphoid expansion and subsequent involution (with control mediated by Foxp3+ Treg)
- IPEX occurs primarily in males (female carriers may have mild disease) and present in early life with diabetes, autoimmune thyroiditis, haemolytic anaemia, eczema, diarrhea, and failure to thrive
- Infections do occur, but immunosuppression is the therapy of choice (using T-cell modulators e.g. cyclosporine)
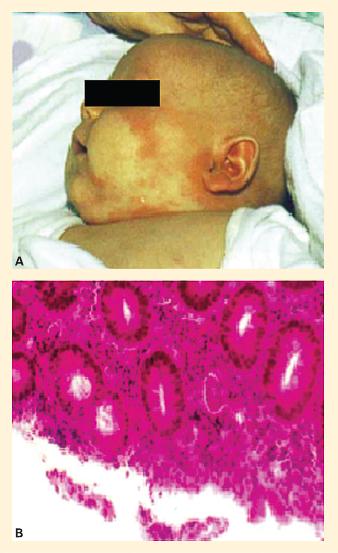
Figure 2. Disease manifestations of FOXP3 deficiency in a child with IPEX syndrome. Panel A: Eczema-like skin lesions on the face. Panel B: Enteritis-like picture in IPEX. Biopsy specimen of the sigmoid colon in a child with IPEX showing a colitis-like picture with infiltration by a mixed cellular infiltrate (hematoxylin and eosin staining). (Reproduced with permission from Foley SC, Préfontaine D, D’Antoni M, et al. Images in allergy and immunology: regulatory T cells in allergic disease. J Allergy Clin Immunol.2007;120:482–6.). [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
Quiz
Related Talk
Kate Webb, Centre for Adolescent Rheumatology – Autoimmune diseases in children



