





Introduction
- Patients with T cell defects can present with a variety of organ specific autoimmune diseases (e.g., type 1 diabetes mellitus in infancy, hypothyroidism, and Addison’s disease) caused by the attack on these organs by the patient’s own immune cells.
- The basis for these clinical complications is unclear, but are thought to be caused by a breakdown in immune tolerance in which a lack of T regulatory cells or the participation of Th17 cells plays a critical role in the pathogenesis of these disorders
Diabetes mellitus type I
- Autoimmune type 1 diabetes mellitus (T1DM) is a disease of undetermined aetiology and mode of inheritance, in which genetically predisposed individuals are exposed to a group of putative environmental exposures that trigger an aggressive and selective autoimmune response against pancreatic beta cells.
- Genetic risk factors involve genes of the MHC (such as DRB1*04, DQB1*02, DQB1*3) and many non-MHC genes (such asPTPN22, CTLA4, IL2).
- Among environmental factors, viral infections (such as CMV and Coxsackie) and vitamin D deficiency are the most documented as causes of TID.
- T1DM is a multi-stage disease characterized by a complex and prolonged autoimmune prodrome (pre-diabetes phase) that develops over months to years and leads to irreversible loss of beta-cell function.
- Several autoimmune markers circulate in the peripheral blood and are readily detectable during the prodrome.
- Three major auto-antigens have been identified in T1DM: insulin, GAD65 and IA2. They were described as major targets for the T lymphocyte attack especially in young children.
- The development of persistent (3 months) single or multiple islet auto-antibodies is thought to occur shortly following killing of beta cells but this seems to occur regardless of insulitis.
- The cellular pathway of the immune system plays a more significant role than the humoral pathway in T1DM; the CD8+ autoreactive T lymphocytes are the most abundant and the most active in beta cell destruction.
- It is proposed that following exposure to a putative antigen, the APCs residing in the islet process and present the auto-antigen to the CD4+ T lymphocytes, driving their activation, as detected by the presence of cell surface activation markers.
- In subjects with insulitis, the islets of Langerhans may be infiltrated by T and B lymphocytes and by monocytes and dendritic cells, supporting a state of chronic inflammation. However, T cells, especially the CD4+ and CD8+ subsets, dominate the insulitis.
- T1DM is primarily a cell-mediated autoimmune disease. Nevertheless, in the absence of specific cellular assays, the early stage of disease is defined by islet autoantibodies.
- The islet autoantibodies therefore represent predictive markers of an ongoing autoimmune response, yet their exact role in beta cell destruction remains to be clarified.
Rheumatoid arthritis
- Rheumatoid arthritis (RA) is a common autoimmune disease affecting approximately 1% of the world’s population.
- RA disease is characterized by a symmetric, polyarthritis of the small joints of the hands and feet, but almost any joint can become involved. The inflammatory process is destructive, and if the patient is not treated, leads to erosion and eventual deformity of the joints (FIGURE 1).
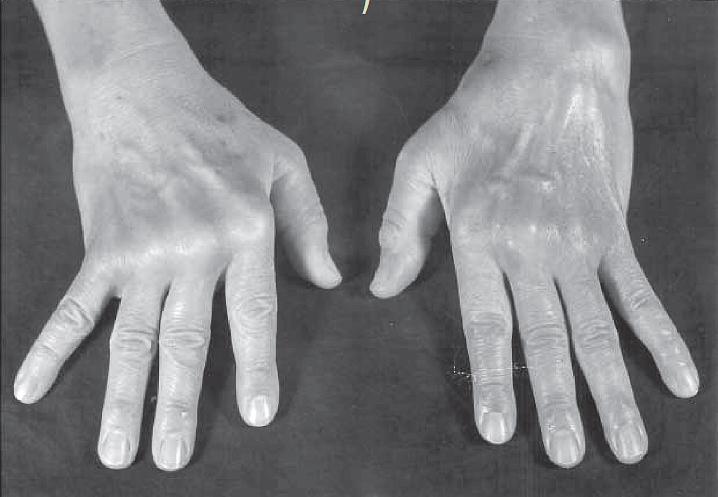
Figure 1: Photograph of the hands of a patient with moderately advanced rheumatoid arthritis. Note the deformities of the metacarpophalangeal joints, with atrophy of the hypothenar muscles and ulnar deviation. (Courtesy of F. Paul Alepa.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- Pathologically, the synovial tissue becomes hypertrophied, highly vascularized, and infiltrated with leukocytes (FIGURE 2).
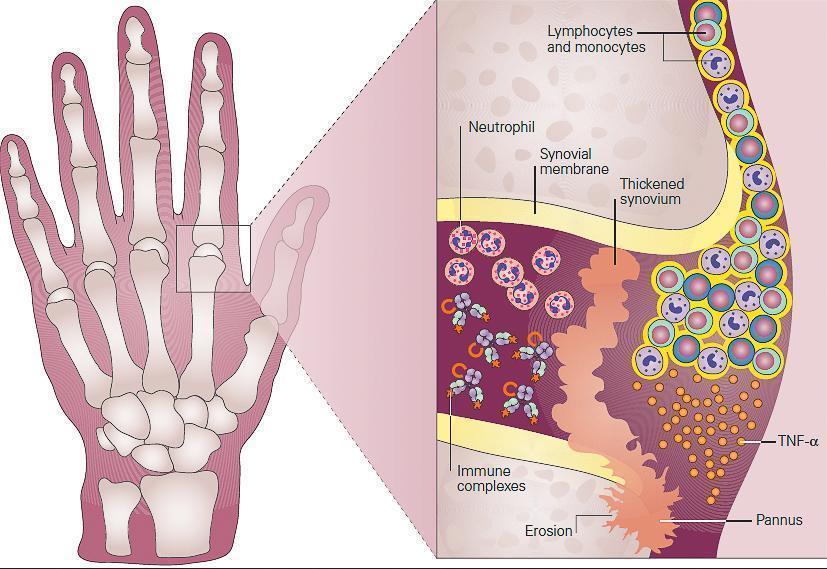
Figure 2: Schematic representation of the role of inflammation in the pathogenesis of rheumatoid arthritis seen in the metacarpophalangeal (MCP) joint of the hand of the patient in Case 2. Shown in the inset, the synovium consists of a synovial membrane and underlying loose connective tissue. In early rheumatoid arthritis, the synovial membrane becomes thickened because of hyperplasia and hypertrophy of the synovial lining cells and begins to invade the cartilage. An extensive network of new blood vessels is formed in the synovium bringing in monocytes and lymphocytes comprised predominantly of CD4+ T cells and B cells (plasma cells) which infiltrate the synovial membrane and synovial fluid. The production of antibody by plasma cells and subsequent formation of antigen-antibody-complement, i.e., immune complexes leads to the chemotactic influx of neutrophils. In established rheumatoid arthritis, the synovial membrane becomes transformed into inflammatory tissue, the pannus, and the production of TNF-α by macrophages and lymphocytes by this invading tissue destroys adjacent cartilage and bone. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- The synovial lining, comprised predominantly of proliferating fibroblasts and infiltrating macrophages, develops into an inflammatory mass, termed the pannus, which can invade and destroy adjacent tissues. The subsynovial area is comprised of infiltrating macrophages and dendritic cells and lymphocytes that are organized into aggregates in a subset of patients.
- The precise cause of RA is unknown but genetic, epigenetic and environmental factors clearly participate:
- Genetic risk factors are prominent, involving genes of the major histocompatibility complex (MHC) as well as many non-MHC genes associated with immune responses and inflammation.
- Among the genetic factors linked to RA susceptibility, several HLA DRB1 alleles (such as DRB1*01, DRB1*04) were reported in different populations and appeared to affect both disease susceptibility and disease severity.
- The finding that all HLA-DRB1 alleles associated with RA risk encode a conserved sequence of 5 amino acids (positions 70–74) that surrounds the peptide-binding pocket of the antigen-presenting molecule led to the ‘shared epitope’ hypothesis.
- Gene-environment interactions have been observed; there is an increased incidence of RA in HLA-DRB1 individuals who smoke cigarettes.
- Changes in a variety of immunologic markers are observed prior to the onset of clinical disease in the sera of patients who develop RA, including anti–citrullinated protein antibodies (ACPA) and rheumatoid factors (RF).
- Of the various inflammatory cells in RA synovium, macrophages have been most clearly implicated in pathogenesis and are the major synovial producers of TNF-a. The role of TNF-a in the pathogenesis of RA deserves special mention as TNF-a blockade has been a major advance in the treatment of RA.
- Currently, TNF- a blockade is commonly used for both acute and maintenance treatment of RA with excellent clinical efficacy.
- Other cells of the innate immune system are also involved. Neutrophils, present in the synovial fluid, synthesize inflammatory prostaglandins, proteases, and reactive oxygen species. Mast cells release cytokines, chemokines, proteases, and vasoactive amines.
- Fibroblast-like synoviocytes (FLS) in the synovial intimal lining also play a key role by producing cytokines that perpetuate inflammation and proteases that contribute to cartilage destruction.
- Rheumatoid FLS develop a unique aggressive phenotype that increases invasiveness into the extracellular matrix and further exacerbates joint damage.
- The observation of T-cell accumulation in the synovium has led to the hypothesis that a T-cell dependent inflammatory reaction to an unknown antigen underlies the pathology.
- Although RA is conventionally considered to be a disease mediated by Th1 cells, attention has increasingly focused on the role of Th17 cells (a subset that produces IL-17A, IL-17F, IL-21, and IL-22 and TNF-α).
Multiple sclerosis
- Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS), resulting in disability.
- The clinical manifestations are very variable and include motor, sensory, visual and cognitive symptoms, none of which are disease specific.
- Neuropathological studies show disseminated patches of demyelination in the brain and spinal cord associated with inflammatory infiltrates containing T lymphocytes.
- A wide range of evidence points to the pivotal role of the immune system in the development of the disease :
- Immune cell infiltrates
- Genetic variants conferring a higher susceptibility to MS which are associated to immune mechanisms (HLA alleles, etc.)
- Autoimmune animal models used for disease characterization and mechanistic studies
- The efficacy of current therapies targeting various components of adaptive immunity.
- The immune dysregulation in MS is considered to be multifactorial, involving genetic susceptibility, epigenetic and post-genomic events, and environmental factors such as viral pathogens, chemicals, smoking, obesity and vitamin D deficiency.
- Over the years, MS has been considered to be an autoimmune disorder where myelin-specific T cells initiate an inflammatory process that results in CNS demyelination.
- These autoreactive T cells are believed to become activated in the periphery and to upregulate adhesion molecules that allow these T cells to interact with and cross the blood brain–barrier and finally establish an inflammatory response directed against myelin.
- The mechanism of activation of these autoreactive, myelin-specific T cells is still not fully understood.
- Processes such as molecular mimicry, where T cells respond to environmental antigens that resemble self-antigens, is a potential mechanism by which these cells get activated.
- For decades, CD4+ T cells have been recognized as playing a major role in the disease, which has led to the development of several therapies. Much of this is due to similarities between MS and its animal model (experimental autoimmune encephalomyelitis; EAE), which is typically induced by CD4+ T cells.
- Strong evidence point to the role of Th1 and Th17 lymphocytes in the pathophysiology of MS.
- The role of CD8+ T cells has recently received additional attention because they are prominent in the inflammatory infiltrate in MS lesions, and have been described to recognize myelin antigens in MS patients, and play a role in breakdown of the blood–brain barrier.
- The role of B cells is MS pathogenesis has also received increased attention. B cells may secrete antibodies that recognize and participate in myelin breakdown.
- The potential importance of B cells in the pathogenesis of MS has been highlighted by studies using the monoclonal antibody rituximab, which recognizes CD20 on B cells and thus results in their depletion.
- Interestingly, newly identified innate-like T cell populations, such as innate lymphoid cells, invariant natural killer T (iNKT) cells, and mucosal-associated invariant T (MAIT) cells, have emerged as important actors in inflammatory diseases.
- Such cells, positioned at the interface between the environment and the host may represent a key link for the amplification of an immune reaction against microbes. Understanding their exact contribution to pathogenesis will undoubtedly open innovative therapeutic possibilities.
Celiac disease
- Celiac disease (CD) is an increasingly prevalent small intestine enteropathy induced by cereal-derived prolamins (dietaray glutens from wheat, rye, barley and sometimes oats) in genetically susceptible individuals.
- Clinical presentation is eclectic. In fact, depending on the extent of intestinal involvement, symptoms may manifest acutely as gastrointestinal pain or malabsorptive diarrhea or more chronically as signs of nutrient malabsorption or as atypical nongastrointestinal manifestations.
- Epidemiological studies show a high prevalence of autoimmune disorders in CD patients and, conversely, a high incidence of CD in autoimmune patients.
- The pathogenesis of celiac disease is predominantly caused by the generation of autoreactive T cells and shares similar characteristics to other autoimmune disorders (FIGURE 3).
- Histological lesions are characterized by the presence of crypt hyperplasia, intraepithelial lymphocytosis, and destruction of the surface epithelial lining of the small intestine.
- The presence of autoantibodies directed against tissue transglutaminase (tTG) suggests that CD has an autoimmune component. Although antibodies to gliadin and tTG are now routinely measured for screening, the gold standard for diagnosis remains the demonstration of villous inflammation or atrophy on small intestinal biopsy.
- The only effective treatment known for celiac disease is avoidance of foods containing gluten. A gluten-free diet has been shown to completely prevent the clinical and pathological complications of celiac disease. Preventing complications and intestinal inflammation is particularly important since, in addition to the manifestations described above, individuals with untreated celiac disease are at increased risk for other serious health conditions, such as autoimmune diseases (i.e., type 1 diabetes mellitus) and certain cancers associated with high mortality, e.g., lymphoma.
- In the current view of the pathogenesis of CD, adaptive immunity plays a key role, accounting for the interplay between the triggering environmental factor, prolamins, and HLA-DQ2/8 haplotypes, the major genetic risk factor.
- Due to their content in proline and glutamine, some gluten peptides can be deamidated by tissue transglutaminase, the autoantibody target, and adopt a configuration that enables their binding into the peptide pocket of HLA-DQ2/8 molecules. Gluten peptides can subsequently be presented to lamina propria CD4 T cells, triggering their activation and the release of interferon gamma (IFN-g).
- It is however now clear that this response, although necessary, is not sufficient and that other factors that impair immunoregulatory mechanisms and/or activate the large population of intestinal intraepithelial lymphocytes (IELs) are necessary for driving tissue damage.
- Strong evidence points to the role of innate immunity orchestrated by the pro-inflammatory cytokine, IL-15.
- In active CD, IL-15, produced by enterocytes and lamina propria dendritic cells and macrophages, favors the activation of IFN-g-producing and cytotoxic CD8+ IELs harboring NK cell receptors.
- Gluten-specific CD4+ T cells may participate in IEL activation via cross-priming or via the production of IL-21 that synergizes with IL-15 to activate cytotoxic CD8+ T cells.
- In the presence of IL-15, CD8-T IELs can induce epithelial lesions via the interactions of their NK receptors NKG2D and NKG2C with their respective ligands MICA and HLA-E, upregulated on enterocytes.
- IL-15 then favors accumulation of activated CD8+ T cells by stimulating their survival and inhibiting their responses to immunoregulatory mechanisms, further increasing the risk of developing T-dependent autoimmune diseases (T1DM, thyroiditis, and perhaps type I refractory celiac disease, RCDI).
- Finally, IL-15 can promote the emergence of unusual IEL-derived T lymphomas sharing characteristics of both NK and T cells and able to drive epithelial lesions. Onset of lymphoma is often revealed by refractoriness to the gluten-free-diet.
- Finally, in active CD, Secretory IgA-gluten complexes formed in the intestinal lumen can bind the CD71 receptor up-regulated at the apical surface of enterocytes, resulting in their rapid retro-transport into lamina propria.
- Binding of SIgA to CD71 may also activate signal transduction into epithelial cells. These mechanisms may exacerbate immune responses.
- Intestinal dimeric IgA can be released into blood and participate in extra-intestinal autoimmunity, notably in dermatitis herpertiformis.
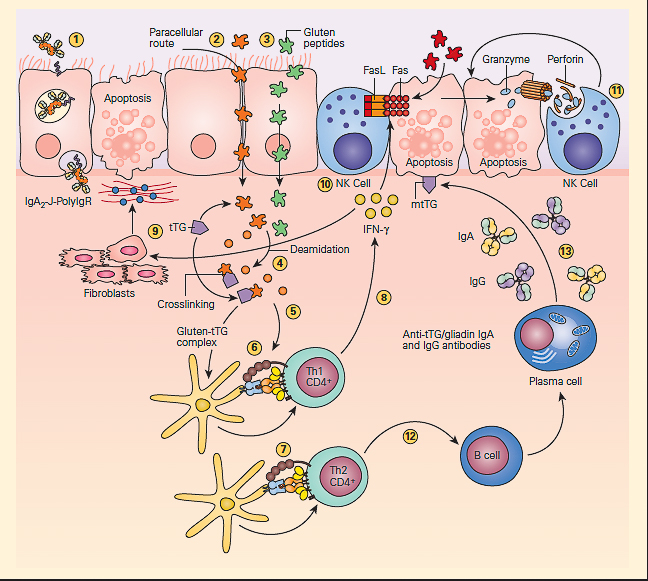
Figure 3: Schematic representation of the pathogenesis of celiac disease. Gluten peptides can be transported across the intestinal epithelium by three main mechanisms: retrotranscytosis of secretory IgA (sIgA) (1) through transferrin receptor CD71 paracellularly (2) or transcytosis as a consequence of impaired mucosal integrity attributable to increased release of zonulin (3). Once internalized, deamidation (4) or cross-linking of gluten (5) by tissue transglutaminase (tTG) leads to the production of deamidated gluten or gluten-tTG, respectively. These gluten peptides are taken up and presented by dendritic cells to either CD4+ Th1 (6) or Th2 (7) cells in the context of HLA-DQ2 or HLA-DQ8 molecules. This presentation leads to activated gluten-reactive CD4+ Th1 cells that produce high levels of proinflammatory cytokines (8), with aTh1 cytokine pattern dominated by interferon gamma (IFN-g). Th-1 cytokines promote inflammatory effects, including fibroblast or lamina propria mononuclear cell (LPMC) secretion of matrix metalloproteinases (MMPs) (9), which are responsible for degradation of extracellular matrix and basement membrane and enhance the cytotoxicity of intraepithelial lymphocytes (IELs) or natural killer (NK) T cells (10). These latter cells facilitate the apoptotic death of enterocytes by the Fas/Fas ligand (FasL) system or interleukin-15 (IL-15)-induced perforin granzyme and NFG2D-MIC signaling pathways (11). Interferon alpha (IFN-α) released by activated dendritic cells further perpetuates the inflammatory reaction by inducing CD4+ T cells to produce IFN-γ. Additionally, through the production of Th-2 cytokines (12), activated CD4+ T cells drive the activation and clonal expansion of B cells, which differentiate into plasma cells and produce antigliadin and anti-tTG antibodies (13). (Adapted with permission from Di Sabatino A, Corazzo GR. Coeliac disease. Lancet. 2009; 373:1480–93.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].



