





- The immune response to cancer is best viewed as a specialised case of immunity in which the malignant cell has adapted and learned how to persist.
- The immunological surveillance theory was originally put forth independently by Burnet and Thomas, who suggested that the immune system continually surveyed the body for the presence of malignant cells, which were continuously arising as a result of mutations.
- Potential strength of immune surveillance is based on the knowledge that cancer cells do indeed express tumour-associated antigens (TAAs) or tumour-specific antigens (TSAs) that can be recognised by the immune system as foreign elements.
- Development of cancer can be explained by the ability of tumour cells to evade immune recognition either by the failure of the immune system to be adequately generated or by the induction of immune tolerance or other inhibitory mechanisms that allows tumour to escape immune detection and elimination (Figure 1).
- In contrast to tumours induced by carcinogens, in which each new tumour has unique antigenic specificity regardless of their morphologic appearance, tumours induced by viruses consistently express antigens that cross-react with other tumours induced by the same or similar viruses even though their morphologic appearance may differ.
Cancer Immunosurveillance versus Immunoediting
- Cancer immunoediting process is envisaged to consist of three phases: elimination, equilibrium, and escape (Figures 1 & 2).
- Elimination phase corresponds to the original concept of cancer immunosurveillance, whereby nascent tumour cells are successfully recognised and eliminated by the immune system, thus returning the tissues to their normal state of function.
- Tumour cells that elude the immunosurveillance phase will progress to the immune editing phase, called the equilibrium phase of advanced oncogenesis, where tumour expansion and metastasis are minimal (tumour dormancy) and usually occur without symptoms.
- In the equilibrium phase, the immune system may eventually eliminate all tumour cells leading to an outcome similar to the elimination phase. In a second scenario, the constant interaction of the immune system with tumours over a long period of time may actually “edit” or sculpt the phenotype of the developing tumour, resulting in the immunoselection of a tumour that has been shaped into a less-immunogenic state.
- Tumours that are no longer susceptible to immune attack then progress into the immunoediting process, termed “escape.” The emergence of clinical symptoms of cancer generally correlates with the escape stage.
- Tumour subverts the immune system, either directly through its nonimmunogenic phenotype, or indirectly through a variety of immunosuppressive mechanisms.
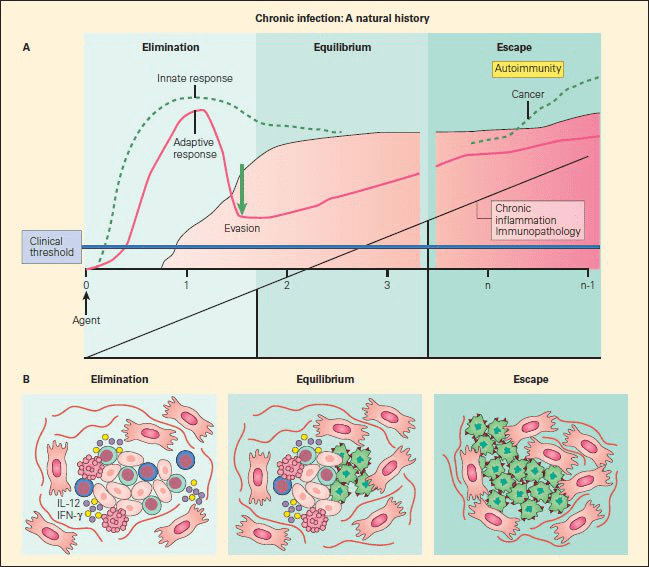
Figure 1. Panel A: Schematic representation of the sequelae of pathogenetic events that follow from failure of elimination of an infectious agent or foreign substance, leading to persistence of the foreign configuration with resultant inflammation, immunopathology in the form of autoimmune disease, or malignancy. Panel B: The three phases of cancer immunoediting: elimination, equilibrium, and escape correlated with the morphologic assessment of emerging cancer cells during these various stages. During the first phase of elimination, only normal cells are seen (red); the few malignant cells that emerge are removed by apoptosis; during equilibrium, both normal cells (red) are seen as cancer cells appear (green); during escape, a predominance of cancer cells are seen (green), with no normal cells evident [Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
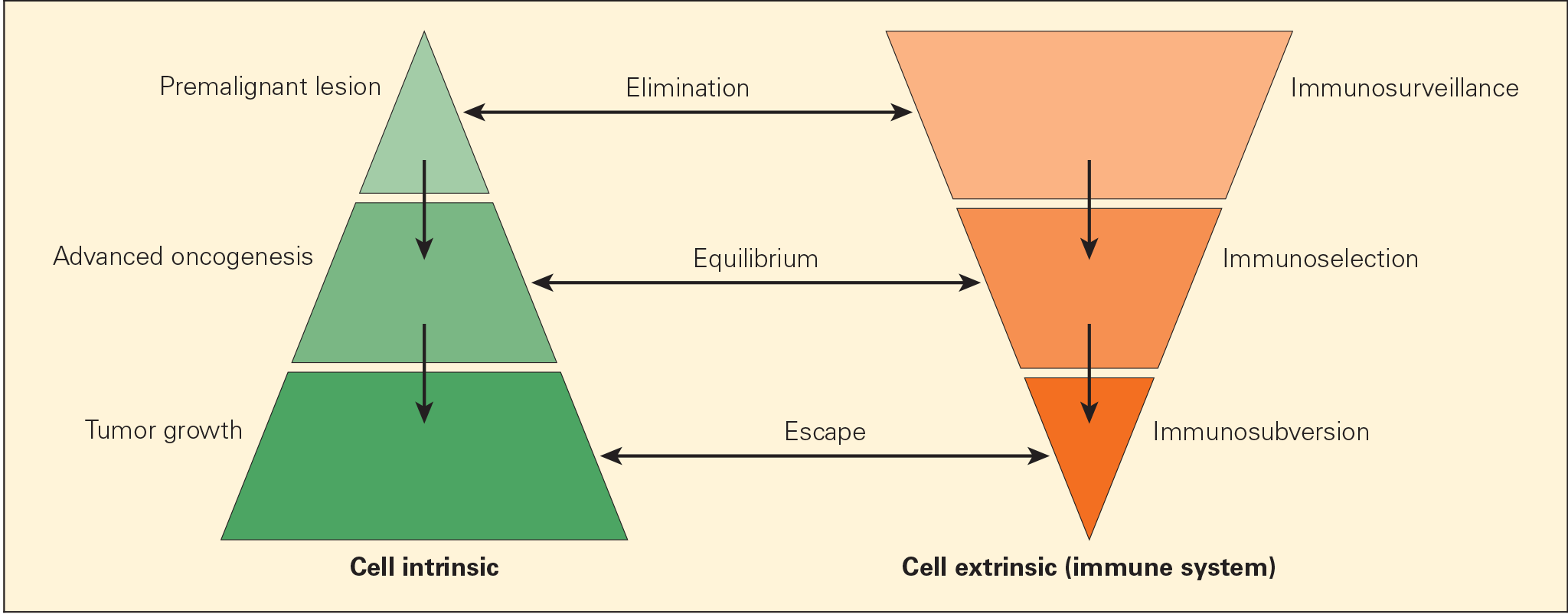
Figure 2. Schematic representation of the immunoediting process consisting of three phases: elimination, equilibrium, and escape, termed the “three Es of cancer immunoediting.” Carcinogenesis is envisaged as a multistep process resulting from cross talk of cancer-cell–intrinsic factors and host immune system (cell-extrinsic) effects. [Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27.]
Clinical Applications of Tumour Antigenicity
- Most responses in tumour immunity are similar to those that apply to allograft rejection phenomena in transplantation models.
- Specific antigens that arise in many tumours are referred to as tumour-associated transplantation antigens (TATAs) or TAAs, which are categorised in four groups: (1) unique antigens specific for an individual tumour; (2) overexpressed peptides or proteins that are found in normal tissue; (3) shared tumour antigens; and (4) viral antigens that may be shared among different cells, but confined to malignancy.
Table 1. Examples of tumour-associated antigens (TAAs) identified in humans
| Category | TAA | Tumour |
|---|---|---|
| Unique tumour-specific antigens | Mutant p21/ras Immunoglobulin idiotype β-catenin Mutant p53 CDK4 Mutant EGFR VIII CEA | Colorectal, pancreatic B cell malignancy Colorectal, breast Pancreatic Melanoma Glioblastoma, lung Colorectal |
| Overexpressed self-antigen peptides | Muc-1 GA733/EpCam Her-2/neu EGF Receptor | Colorectal Colorectal Breast Colorectal, lung, head, and neck |
| Shared tumour antigens | Melanoma antigen E (MAGE) tumour-associated antigen | Melanoma |
| Viral-associated antigens | Human papilloma virus (HPV) Hepatitis B virus (HBV) Epstein–Barr virus (EBV) | Cervical Hepatocellular B cell malignancy |
Table 1. Examples of tumour-associated antigens (TAAs) identified in humans. [Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
- In vitro and in vivo studies suggest that antitumour CD8+ cytotoxic lymphocytes (CTL), ADCC-mediated natural killer (NK) cells, or antitumour antibodies can participate in tumour destruction and tumour regression.
The Role of the Innate Immune System in Tumour Immunity
- More recently it has become apparent that innate immune responses may also play an important role in resistance against the development and progressive growth of tumours.
- Macrophages and mast cells can activate vascular and fibroblast responses in order to orchestrate the elimination of the malignant cell and to initiate local tissue repair.
- DCs, on the other hand, take up tumour antigens and migrate to lymphoid organs, where they present processed peptides to T cells for the induction of specific antibody and CMI responses.
- NK cells also participate in cellular cross-talk between innate and adaptive immune cells through their ability to interact bidirectionally with DCs. That is, certain NK cell subsets eliminate immature DCs, whereas others promote DC maturation, which can also reciprocally regulate activation of NK cells.
- Induction of efficient primary adaptive immune responses requires direct interactions with mature antigen-presenting cells and a strong pro-inflammatory milieu.
The Role of the Adaptive Immune System in Tumour Immunity: Recognition of TAAs on Tumour Cells by T Cells and Other Cells
- TAAs comprise short amino acid peptide segments, which might be derived from any intracellular protein.
- T cells recognise these TAAs through their TCRs in the context of MHC-I or MHC-II on the surface of tumour cells or APCs, respectively.
- Two distinct pathways have been identified for the processing of TAAs, the exogenous and endogenous pathways.
- In the endogenous pathway, tumour cells continually degrade unfolded intracellular proteins within the proteosome into short peptide fragments. Following transport through several pathways in the endoplasmic reticulum, these fragments are then loaded onto the MHC-I. The final MHC-I/peptide complexes are then transported to the tumour cell surface through the Golgi apparatus for presentation to CD8+ T cells (Figure 3).
- In the exogenous pathway, APCs, through a variety of endocytic pathways, take up intracellular proteins that have been released from damaged or injured tumour cells. These intracellular proteins can then be degraded in lysosomal pathways to peptides that, when complexed with MHC-II on the cell surfaces, are presented to CD4+ T cells.
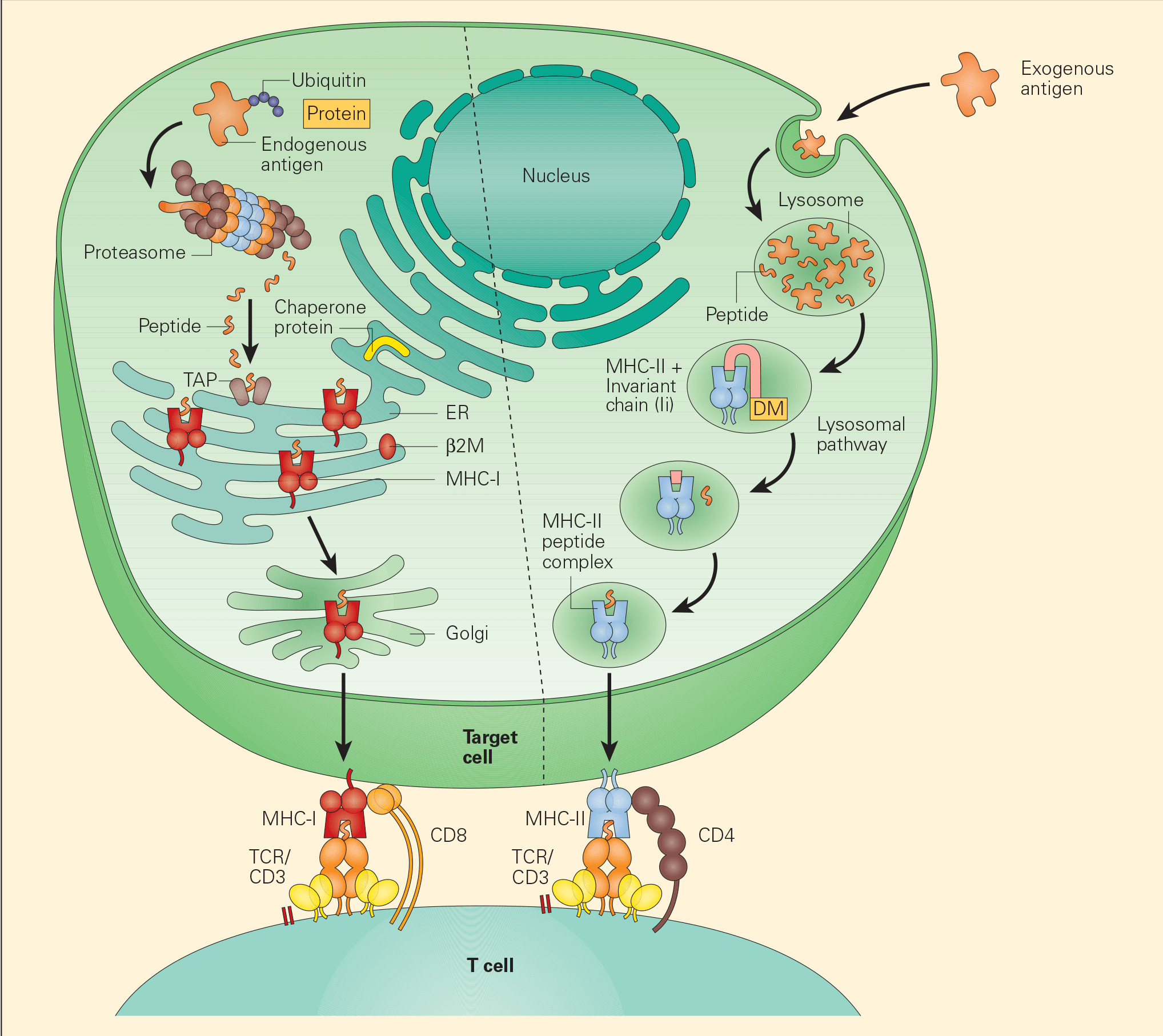
Figure 3. Tumour-associated antigen processing. Abbreviations: ER = endoplasmic reticulum; MHC = major histocompatibility complex; TCR = T cell receptor. [Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- Alternatively, APCs may also process the tumour proteins through the endogenous pathway. In this way, APCs are able to prime both MHC-I and MHC-II responses and give rise to specific antibody and cell-mediated immune responses important in tumour immunity.
- The MHC-II/peptide complexes expressed on the surface of APCs and presented to naïve T cell CD4+ helper cells are followed by further maturation into T helper 1 (Th1), Th2, Th17, and Treg populations that function to promote delayed hypersensitivity, antibody production (through B cell interaction), inflammation, or immunosuppression, respectively.
- Recent studies have shown the active participation of Th17 in tumour immune responses (Figure 4).
- The CD8+ cytotoxic T cells that recognise the MHC-I/peptide complex expressed on tumour cells, on the other hand, result in tumour cell lysis and apoptotic cell death.
- The recognition of peptide-MHC complexes by T cells through their TCRs allows the immune system to discriminate those tumour antigens that are distinct from self antigens, as the latter have either induced deletion of self-recognised T cells or developed tolerance. This development of tolerance is now considered a prime mechanism underlying immune evasion by cancer cells and therefore a prime target for immune intervention.
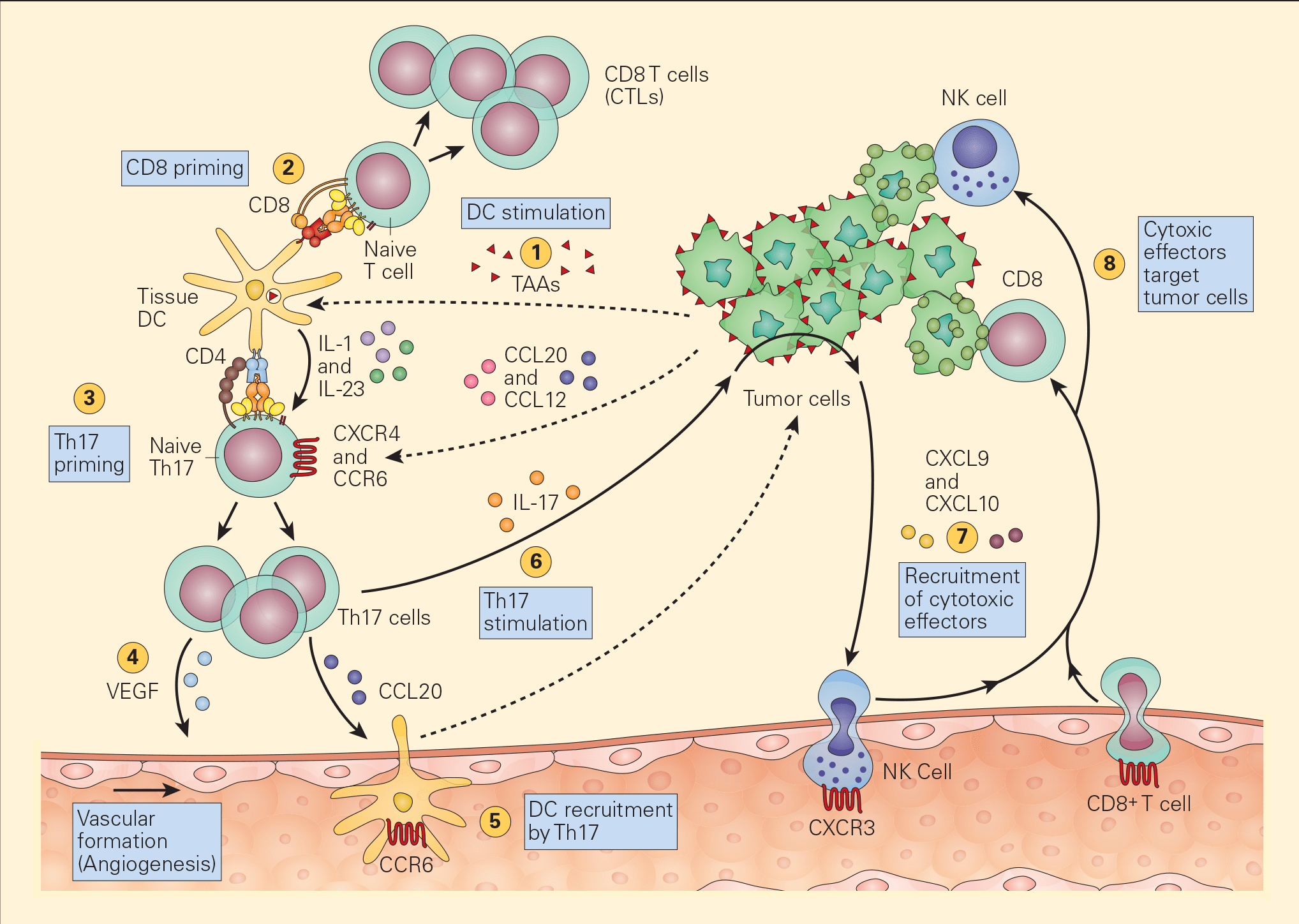
Figure 4. Schematic representation of the various pathways that Th17 cells play in antitumour immunity. Tumour-associated antigens (TAAs) shed from tumour cells are taken up by resident tissue dendritic cells in the tumour microenvironment (1) and following cognate presentation of MHC-I/peptide or MHC-II/peptide to CD8 (2) or Th17 cells (3), respectively, there is clonal expansion of antitumour CTLs as well as Th17 activation that secretes angiogenic factors, i.e., VEGF (4) and chemotactic factors for the recruitment of DCs (5). The stimulation of Th17 cells and their synthesis of IL-17 (6) induces the tumour cells to secrete chemotactic factors, e.g., CXCL9 and CXCL10 (7), leading to the recruitment of additional cytotoxic effectors, i.e., NK and CD8+ cells. [Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Cross-presentation and Cross-priming Tumour Antigens
- T cell activation requires two distinct signals. Signal 1 is delivered by the interaction between the T cell receptor (TCR) and the antigenic peptides presented on MHC molecules. Signal 2 is provided by one of several nonspecific co-stimulatory molecules such as the binding of CD28 on T cells with B7 family molecules on antigen-presenting cells (APCs).
- Cancer cells frequently stimulate Signal 1 alone, and inefficiently Signal 2. Accordingly, cancer cells preferentially induce tolerance.
- Tumour antigens must be presented by antigen-presenting cells to initiate and sustain anti-tumour immune responses. This is achieved by a process called cross-presentation, namely, tumour cells or tumour antigens are taken up by APCs, which process the antigens and present them on the APC cell surface restricted on MHC-I and MHC-II molecules.
- Together, signal 1 and signal 2 activate naïve T cells in a process called cross-priming. This process can also cause T cell unresponsiveness or cross-tolerance.
Cytotoxic T- and NK- Cancer Cell Killing
- After generation of mature CTL cells resulting from the APC-antigen-T cell interaction, the presence of the tumour peptide-loaded MHC-I molecule on the surface of the cancer cell is required for effective tumour cell killing.
- During the course of malignant transformation of a normal cell to a malignant cell, cancer cells may lose the MHC-I molecule on its cell membrane as part of its evasion strategy to elude its destruction by the CTL.
- In the event of the loss of MHC-I expression by cancer cells, NK cell interaction with them can only occur through killer-activating receptor (KAR) ligand, resulting in their killing.
- An alternative mechanism of destruction of tumour cells by NK cells can occur by an ADCC mechanism where the Fab portion of an IgG antibody produced by B cells binds to the surface TSA and bridges to an Fc receptor on the NK cell.
The Role of Treg Cells in Tumour Immunity
- Regulatory T cells are thought to be recruited in the periphery by the tumour cells themselves.
- Regulatory T cells functionally suppress immune responses by influencing the activity of another cell type and can occur either by cell-to-cell contact or by the elaboration of immune-regulatory cytokines IL-10 and TGF-β.
- T cells comprise a variety of subsets that are conceptually divided into three populations: (1) the classic regulatory CD4+CD25+FOXP3+ T cells are thought to be thymus derived, and are termed naturally occurring regulatory T cells (nTreg cells); (2) CD4+IL-10+FOXP3- regulatory T cells that are induced in vitro with various protocols or in vivo in response to exogenous antigen challenge and are termed adaptive regulatory T cells, induced regulatory (iTreg) T cells, or T regulatory 1 cells (TR1 cells); and (3) CD4+TGF-β+ T cells that are also induced Treg cells activated in the periphery in the context of oral tolerance and are termed TH3 cells.
- Regulatory T cell-mediated immune-suppression is one of the crucial tumour immune-evasion mechanisms and the main obstacle for a successful tumour immunotherapy.
- Tregs also play an important role in suppressing the effector mechanisms directed against TAAs resulting from the myriad of factors produced within the tumour microenvironment.
- The most common postulated mechanism of Treg cell incitement is by the tumour expression of chemokines, such as CCL22, that bind to specific chemokine receptors, such as CCR4, on Treg cells. (Figures 5 & 6).
- Dysfunctional APCs induce Treg cell activation and expansion into more powerful suppressive phenotypes. Moreover, the presence of TGF-β itself converts CD4+CD25- T cells into iTreg cells by their synthesis of CD25 and FOXP3.
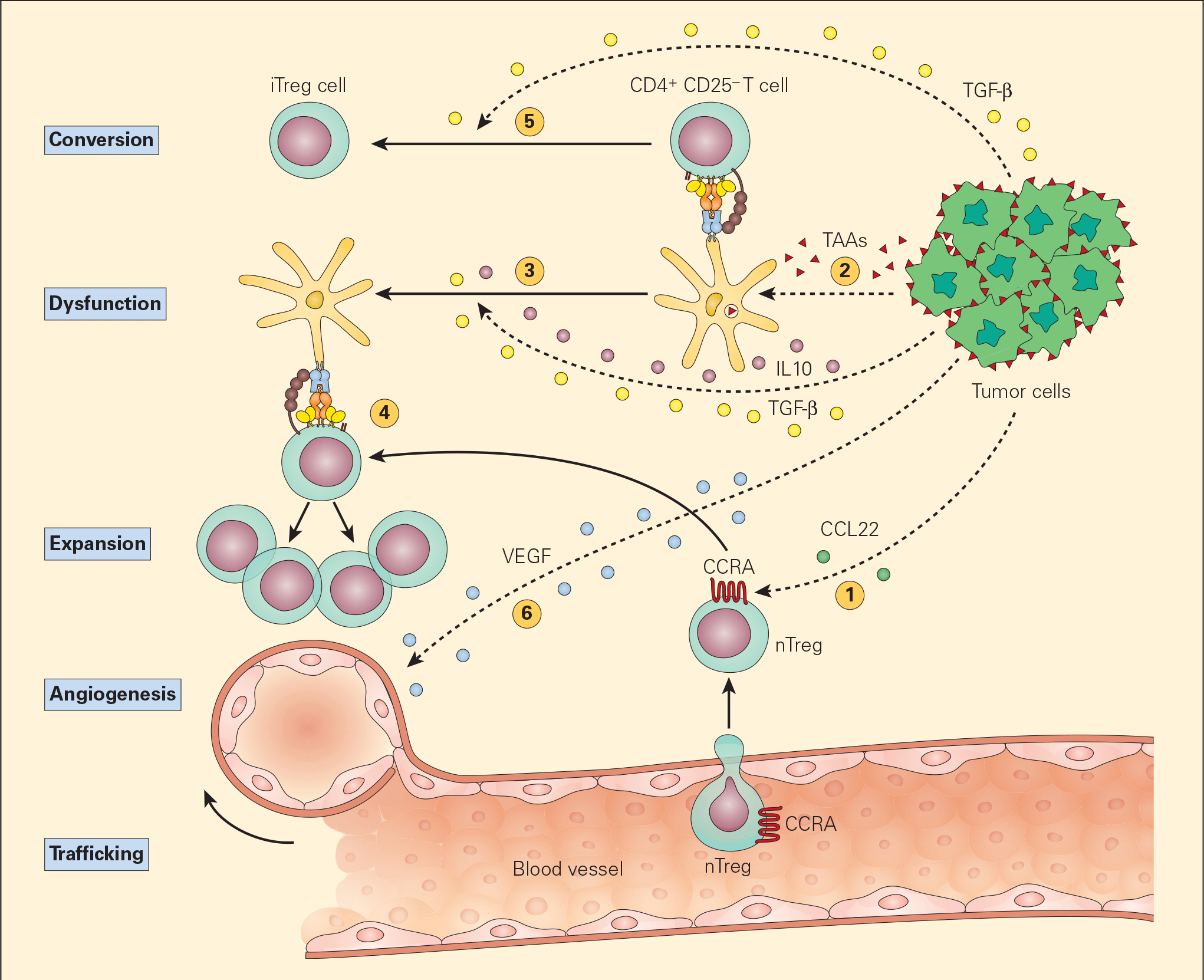
Figure 5. Schematic representation of the various mechanisms that the tumour microenvironment uses in suppressing the antigen-presenting capacity and function of resident DCs. Following tumour production of chemokines (e.g., CCL22), nTreg cells are expressing CCR4 and are recruited from the vascular compartment (1). Following their release from the tumour, TAAs are normally taken up by DCs (2) leading to the activation of CD4+CD25- T effector cells. The subsequent release of the immunoregulatory cytokines IL-10 and TGF-β from the tumour suppresses DC differentiation and function, resulting in a dysfunctional DC (3). The nTregs recruited by the tumour are stimulated by the dysfunctional DCs to expand (4) leading to immunosuppressive activity in the tumour environment. The TGF-β released from the tumour cells can also lead to the conversion of the CD4+CD25- T effector cells into CD4+CD25+ Treg cells, leading to further immunosuppressive activity (5). The tumour production of VEGF stimulates angiogenesis. [Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
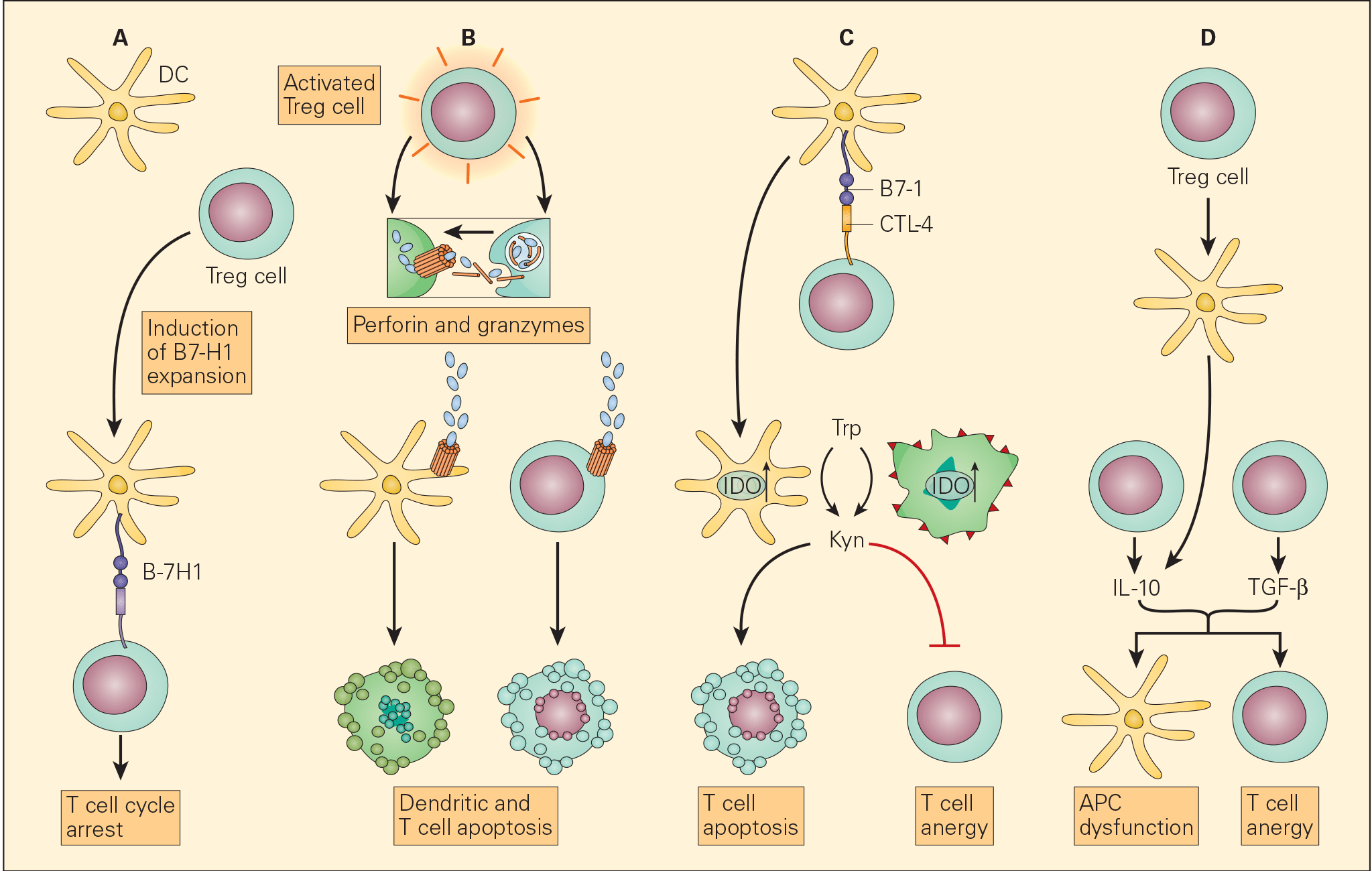
Figure 6. Schematic representation of the various proposed mechanisms of the immunosuppressive action of Treg cells in the tumour microenvironment. Panel A: The induction of APCs to express B7-H4 by Treg cells leads to suppression of T effector cells. Panel B: The enhanced perforin/granzyme B pathway by activated Treg cells may also induce cytotoxicity of DCs and effector T cells. Panel C: The interaction of CTLA-4 with CD80/CD86 on APCs, Treg cells can exert their immunosuppressive effects by the induction of indoleamine 2,3-dioxygenase (IDO) that favors T cell anergy and/or apoptosis. Panel D: An increased concentration of TGF-β produced both by the Treg cells as well as the tumour cells leads both to APC dysfunction and T cell anergy. [Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006; 6: 295-307.]
Role of Antibodies in Tumour Immunity
- In addition to their indirect beneficial role in the ADCC mechanism of tumour killing, antibodies can also play a direct role in tumour immunity.
- Antibody directed to TAA together with activation of the complement system can kill the cancer cell by cytolytic activation of the membrane-activation complex (MAC) cascade.
- Alternatively, TAA alone or as antigen-antibody complexes can interfere with the CTL killing of a cancer cell by blocking its activity.
Mechanisms of Tumour Cell Evasion of the Immune Response
- A number of potential mechanisms may underlie the capacity of a malignant cell to avoid destruction at the hands of the immune system.
- The countermeasures that a tumour may employ include down regulation of potential target antigens and inhibition of the T cell response and direct modulation of proinflammatory cytokines. These evasion mechanisms may serve as novel targets for cancer therapy.
- One of the many factors that promote immune escape and tumour growth in cancer cells is activation of the enzyme indoleamine 2,3-dioxygenase (IDO). IDO is an enzyme that converts tryptophan (Trp) to kynurenine (Kyn) and is involved in tumour growth and immune suppression (Figure 7).
- IDO is an immunomodulatory enzyme produced by some alternatively activated macrophages and other immunoregulatory cells that catalyses tryptophan through the generation of kynurenine and has been associated with mechanisms of cancer escape and expansion. Originally, activated Treg cells would induce the expression of IDO in DCs that will translate signals promoting T cell anergy and apoptosis. This state of anergy would be expanded by the arrival of these dysfunctional DCs expressing high cytoplasmic concentration of IDO into the draining lymph nodes. Their subsequent interaction and suppression of local effector T cells, like CTL, would reduce immunosurveillance in the lymph node favouring uncontrolled metastasis of tumour cells to these converted sites. Additionally, IDO has been linked to the inactivation of the tumour suppressor gene Bin1 that has been implicated in the genesis of uncontrolled growth in cancer cells. So, IDO plays a dual role favouring cancer; it promotes cancer growth and suppresses immunosurveillance.
- The clinical implications of this metabolic pathway has led to the development of small molecule therapeutic inhibitors of IDO that can be used to augment the efficacy of traditional chemotherapeutic drugs used to treat various forms of human cancer.
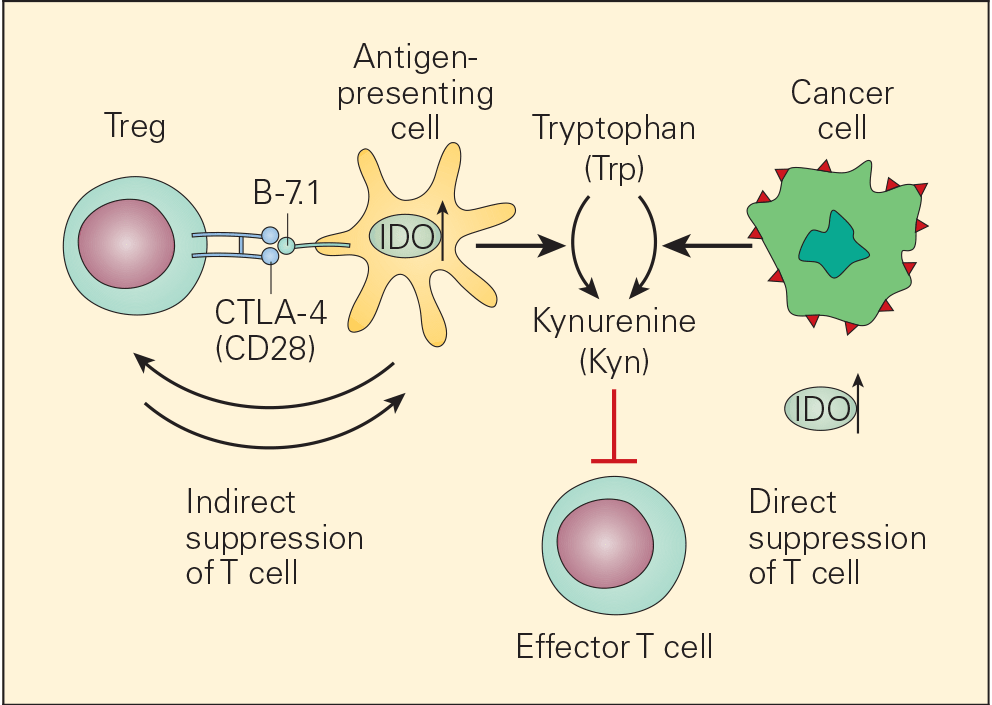
Figure 7. Schematic representation of the dual roles of IDO in cancer. IDO is an important immunomodulatory enzyme produced both by activated macrophages as well as by other immunoregulatory cells and performs its normal immunoregulatory functions by catalyzing the conversion of tryptophan to kynurenine. By consumption of tryptophan and overproduction of kynurenine, IDO upregulation leads to T cell suppression both by blunting T effector cell function as well as recruitment of Treg cells. IDO is also upregulated in cancer cells where it is thought to contribute to the immune subversion strategy used by many tumours. IDO dysregulation may exert its tumour promoting effects through its known ability to inactivate the tumour suppressor gene Bin1 as well as by incapacitating effective immune tumour surveillance mechanisms, thereby promoting tumour growth. [Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Proinflammatory Cytokines Involved in Tumour Cell Evasion of the Immune Response
- Tumour necrosis factor-alpha (TNF-α) and lymphotoxin (LT-α or TNF-β) are two interrelated members of the TNF family. While LT-α is predominantly produced by lymphocytes, TNF-α is produced by macrophages, lymphocytes, and other cells in selected situations.
- The in vitro effects of these cytokines include growth inhibition or lysis of transformed cells, activation of phagocytic cells, upregulation of growth factors, and control of development and expression of cell-mediated antitumour immune responses.
- They also display a wide spectrum of reactions in vivo, some of which include necrosis of tumours, leukocytosis and inflammation, cachexia, and shock. Their biologic effects are induced by their engagement with their specific cell surface receptors.
- Two clinically important TNF receptors are TNF-RI (55 kD), expressed by most cell types, and TNF-RII (75 kD), restricted to lymphoid cells. These two distinct families of TNF receptors are found either on cell surfaces or as soluble TNF Receptors (sTNF-Rs).
- These sTNF-Rs are expressed constitutively and, when released by the malignant cell, are thought to be another form of tumour evasion by their capacity to bind to TNF-α and to inhibit its activity in the surrounding microenvironment of an immune target.
- The removal of soluble cytokine inhibitors present in the plasma of patients with a variety of cancers, might, therefore, be a unique therapeutic intervention leading to tumour regression by increasing local tumour destructive inflammatory responses while sparing systemic effects.
- The overproduction of TNF-α (originally called “cachexin”) and other proinflammatory cytokines may also be the mechanism for the systemic effects of fever, weight loss, malaise, and cachexia seen in cancer patients with extensive disease.
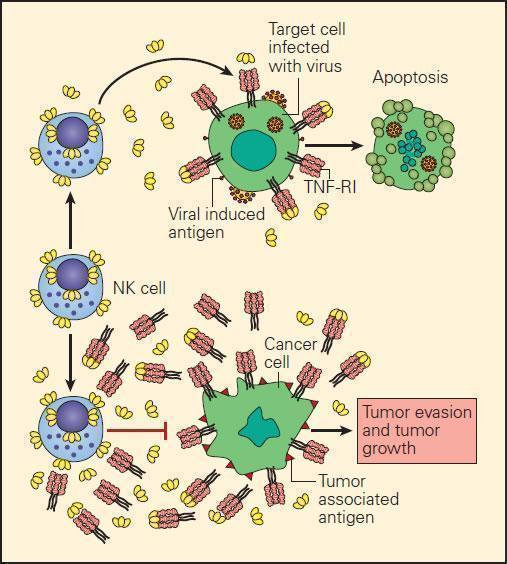
Figure 8. Schematic representation of the proposed normal killing mechanisms of a tumour cell by a TNF-producing activated CTL compared to inhibition of TNF killing resulting from its neutralisation by excessive production of s-TNF-Rs in the surrounding microenvironment of the tumour cell. [Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
General Concepts of the Role of Inflammation in Tumour Immunity or Progression: Beneficial or Detrimental Outcomes
- There is accumulating evidence to suggest that a perturbed innate/adaptive immune balance as seen in chronic inflammation or chronic infection may also enhance conditions for tumourigenesis.
- Genetic predisposition underlies some disorders, such as pancreatitis, ulcerative colitis, and some rheumatoid diseases.
- Others are associated with infectious disease pathogens. For example, Helicobacter pylori causes chronic gastritis in infected hosts and in some patients may be associated with gastric cancer, e.g., adenocarcinoma and lymphoma, whereas infection with hepatitis B or hepatitis C virus (HBV and HCV, respectively) is linked to chronic hepatitis, cirrhosis, and in some patients with subsequent development of hepatocellular carcinoma. Similarly, infection with HPV has been associated with vulvar squamous cell carcinomas and adenocarcinomas.
- Unresolved inflammation resulting from exposure to toxic factors such as asbestos or smoking, as well as from ongoing chemical or physical irritation, such as acid-reflux disease or exposure to ultraviolet (UV) light, may therefore be related to the development of lung cancer, gastroesophageal junction cancer, and skin cancer, respectively.
- Mutations and/or genetic polymorphisms in crucial genes that regulate cytokine function, metabolism, and leukocyte survival have also been implicated as aetiological factors in chronic inflammation, thus lending further support for the possible relationship of chronic inflammation and cancer.
- During acute inflammation, innate immune cells, including phagocytic cells and NK cells, form the first line of immune defense and regulate subsequent activation of adaptive immune responses. During chronic inflammation, these roles can be reversed—i.e., adaptive immune responses can cause ongoing and excessive activation of innate immune cells.
- Whereas acutely activated innate immune cells contribute to efficient T cell activation, chronically activated innate immune cells can cause T cell dysfunction through the production of reactive oxygen radicals.
- Regardless of the underlying initiating cause or pathogenetic mechanism, if an infectious or assaulting agent is inadequately cleared and persists in tissue, or a tissue is subjected to ongoing insult and damage that fails to heal in a timely manner, host inflammatory responses can persist and exacerbate chronic tissue damage.
- Failure of elimination of a foreign substance, may lead to a chronic process, i.e. further malignant progression and cancer expansion. It may now be possible to superimpose the “three Es of cancer immunoediting,” i.e., elimination, equilibrium, and escape, upon the three progressive stages of the immune response evolving from innate and adaptive immunity and terminating in the chronic irreversible phase of cancer.
Myeloid-Derived Suppressor Cells
- Under pathological conditions such as infection, trauma, immunosuppression, or autoimmunity, a partial block in immature myeloid cells (IMC) differentiation results in the abnormal expansion of myeloid-derived suppressor cells.
- Moreover, those pathologic conditions may favour their differentiation into myeloid-derived suppressor cells (MDSCs) expressing STAT-induced immune suppressive factors, such as arginase 1, inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS), with a reduced expression of surface cell molecules like MHC-I and a consequent suppression of antigen-specific T cell activation.
- In response to tumour derived factors, MDSCs have exhibited the capacity to differentiate into tumour-associated macrophages (TAMs) distinguished by the absence of GR1 concurrent to the absence of any of the IMC-specific proteins such as arginase 1 or iNOS, depending on tumour microenvironment.
- TAMs contribute to antigen-non-specific-T cell suppression in cancer and are capable of producing IL-1β, IL-6, IL-10, and TGF-β.
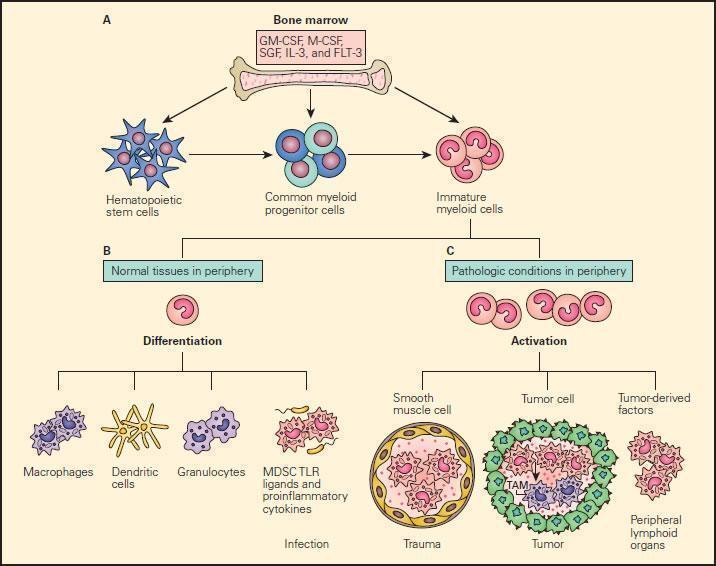
Figure 9. Schematic representation of the normal and pathological differentiation of myeloid cells. Panel A: In the bone marrow, hematopoietic stem cells can progressively differentiate into common myeloid progenitor and then to immature myeloid cells under the inductive influence of several growth factors, including GM-CSF, M-CSF, SCF, IL-3, and FLT-3. The immature myeloid cells migrate into peripheral tissues where they can differentiate and participate in either physiologic or pathologic conditions according to the local environment. Panel B: Shows the normal differentiation of myeloid precursors into macrophages, dendritic cells, granulocytes, and myeloid-derived suppressor cells (MDSCs) in peripheral tissues in response to infection. Panel C: Shows the progressive changes of trauma, tumour development peripheral lymphoid infiltration induced by tumour derived factors. In response to tumour-derived factors, MDSCs exhibit the capacity to differentiate into tumour-associated macrophages (TAMs) and contribute to antigen-non-specific T cell suppression in cancer. SCF = stem cell factor; FLT3 = FMS-related tyrosine kinase 3. [Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74.]
Role of Innate Lymphoid cells in Cancer
- NK cells were long considered the only lymphocytes of the innate immune system. However, in recent years, have been identified novel cell subsets of lymphoid origin, the innate lymphoid cells (ILCs), which lack rearranged antigen-specific receptors, like TCR and BCR.1.
- ILCs are required for the formation of secondary lymphoid organs, development of mucosal-associated lymphoid tissues and serve as a rapid and early source of the effector cytokines during infections. 1, 2
- ILCs differentiate from common lymphoid progenitors (CLP) giving rise to committed ILC progenitors (ChILP).1,2 , that differentiate into cytotoxic ILCs, such as conventional NK (cNK) cells, and helper-like ILCs, such as the ILC1, ILC2, and ILC3 subsets. 1
- Every ILC group share with an adaptive counterpart some of the transcriptional factors governing their differentiation and the cytokine profile they produce.1,3
- Cytotoxic ILCs share characteristics with CD8+T cells, including the expression of the transcription factors Eomes and T-bet and the production of IFN-γ .1,2
- IFNγ have cancer killing properties, is capable of inhibiting cell proliferation, stimulates NK cells and up regulates the expression of MHC class I and II molecules on tumour cells, as well as the adhesion molecules and transcription factors in T cells that mediate these lymphocytes’ tumour killing properties.1,2 . IFNγ also induces the expression of IP10 and MIG chemokines in endothelial cells, both of which act to inhibit tumour angiogenesis.2
- ILC2s are dependent upon the expression of GATA3 and, like Th2 cells, secrete IL-5, IL-13 and the epidermal-growth-factor-like molecule amphiregulin to control helminth infections.2 . Their role in cancer surveillance is uncertain, but it has been suggested that ILC2s promote the growth and metastasis of breast cancer cells partially mediated by IL-33/IL-13 pathways.
- In contrast, studies in metastatic melanoma have shown that ILCs derived IL-5 (activated by IL-33) may play an important role in tumour surveillance by recruiting lung eosinophils.
- RORγt+ILC3s correspond to Th17 cells. In mice, expression of the chemokine receptor CCR6 distinguishes CCR6+ILC3s and CCR6−ILC3s, which both produce IL-22. The CCR6+ILC3 population comprises lymphoid-tissue-inducer cells (LTi) cells and LTi-like cells. In addition to secreting IL-22, CCR6+ ILC3s secrete IL-17.2. Excessive IL22 production is observed in some cases of hepatic carcinoma and has been associated with tumourigenesis and tumour growth, the inhibition of apoptosis, and the promotion of metastasis due to STAT3 activation.2
- Patients with inflammatory bowel disease show an accumulation of RORγt+ ILC3s in the intestine where may be playing key roles in the pathogenic inflammation gut mucosa. ILC3s appear to be able to induce colorectal cancer through the IL-23/IL-17 signaling pathway in the presence or independently on gastrointestinal infection.1,2
- Finally, LTi cells are apparently involved in immunotolerant melanoma expressing the chemokine CCL21, although more research is needed to comprehend the biological functions of ILCs in normal and malignant processes.
Quiz
References and Links
- Vallentin, B. et al. Innate Lymphoid Cells in Cancer. Cancer Immunol. Res. 3, 1109–14 (2015).
- Tian, Z., van Velkinburgh, J. C., Wu, Y. & Ni, B. Innate lymphoid cells involve in tumorigenesis. Int. J. Cancer 138, 22–29 (2016).
- Klose, C. S. N. & Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 17, 765–774 (2016).
- Gajewski, T., Schreiber, H. & Fu, Y. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunology. 14,1014–1022
- Palucka, A. & Coussens, L. The Basis of Oncoimmunology. Cell. 164: 1233–1247 (2016)
- Kepp, O. et al. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014; 3(9): e955691. (2014)
- Cohen, J. et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. The Journal of Clinical Investigation. 125(10):3981-3991. (2015)
- & J Immunol. 195:5117-5122. (2015)
- Klebanoff, C., Rosenberg, A, & Restifo, N. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature Medicine. 22,26–36.
- Strønen, E. et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 10.1126. (2016)
- Tran, E. et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science. 344:641-645. (2014)



